
ABSTRACT
The public's health is gravely at risk due to the current global outbreak of emerging viruses, specifically SARS-CoV-2 and MPXV. Recent studies have shown that SARS-CoV-2 mutants (such as Omicron) exhibit a higher capability to antagonize the host innate immunity, increasing their human adaptability and transmissibility. Furthermore, current studies on the strategies for MPXV to antagonize the host innate immunity are still in the initial stages. These multiple threats from emerging viruses make it urgent to study emerging virus-host interactions, especially the viral antagonism of host antiviral innate immunity. Given this, we selected several representative viruses that significantly threatened human public health and interpreted the multiple strategies for these viruses to antagonize the host antiviral innate immunity, hoping to provide ideas for molecular mechanism research that emerging viruses antagonize the host antiviral innate immunity and accelerate the research progress. The IAV, SARS-CoV-2, SARS-CoV, MERS-CoV, EBOV, DENV, ZIKV, and HIV are some of the typical viruses. Studies have shown that viruses could antagonize the host antiviral innate immunity by directly or indirectly blocking antiviral innate immune signaling pathways. Proviral host factors, host restriction factors, and ncRNAs (microRNAs, lncRNAs, circRNAs, and vtRNAs) are essential in indirectly blocking antiviral innate immune signaling pathways. Furthermore, via controlling apoptosis, ER stress, stress granule formation, and metabolic pathways, viruses may antagonize it. These regulatory mechanisms include transcriptional regulation, post-translational regulation, preventing complex formation, impeding nuclear translocation, cleavage, degradation, and epigenetic regulation.
Introduction
The recent global outbreaks of SARS-CoV-2 and MPXV have had a significant impact on public health security worldwide. Recent data have shown that multiple factors contribute to the severity of COVID-19. Severe COVID-19 patients also had downregulated ISGs and decreased interferon production [Citation1]. Notably, since its discovery in November 2021, the Omicron mutant strain has spread rapidly to about 150 countries and regions worldwide. Studies have found that Omicron, the most severe variant of SARS-CoV-2 so far, has evolved a greater ability to antagonize the host innate immunity, thereby increasing its human adaptability and transmissibility [Citation2]. Monkeypox cases have been recorded nonstop since May 2022 in a number of non-African nations and areas. Monkeypox is a zoonotic disease caused by MPXV. More than 80,000 instances of monkeypox have been reported worldwide to date, according to the World Health Organization, in roughly 112 nations and territories. Of those cases, 116 have been deadly [Citation3].
The public's health is at risk from a number of viruses and linked infectious diseases in addition to SARS-CoV-2 and MPXV. One common respiratory pathogen is the influenza virus. Seasonal influenza outbreaks kill between 250,000 and 500,000 people worldwide each year [Citation4]. There are four types of influenza viruses: A, B, C, and D. Among all subtypes, IAV is the most common [Citation5]. Besides, highly pathogenic coronaviruses also include SARS-CoV and MERS-CoV, which have been transmitted to humans and caused severe acute respiratory illness over the past two decades [Citation6,Citation7]. HIV-1 continues to be a major global public health concern as of right now, with over 40 million people infected and 20 million cases resulting in death worldwide [Citation8]. Notably, these continuous outbreaks of infectious diseases caused by EBOV, DENV, and ZIKV have also caused significant threats to global health in the past few years. Between 60% and 90% of people who contract EBOV die from a severe hemorrhagic illness [Citation9–11]. Furthermore, DENV is transmitted to humans via mosquitoes, resulting in about 400 million infections and tens of thousands of deaths yearly [Citation12,Citation13]. ZIKV has infected millions of individuals in the past ten years by causing significant epidemics in the tropics and spreading to other areas [Citation14–17].
Researching the relationships between emerging viruses and hosts, particularly the viral antagonistic relationship with host antiviral innate immunity, is crucial given the numerous risks posed by emerging viruses such as SARS-CoV-2 and MPXV. We can only have a more comprehensive understanding of emerging viruses and their pathogenic mechanisms to quickly respond to each outbreak of mutant strains and global epidemics by accelerating the study's progress on emerging viral antagonism of host antiviral innate immunity. The mechanisms by which viruses antagonize the host antiviral innate immunity has been the subject of countless investigations over the last ten years, which have resulted in the formation of a rather organized theoretical framework. In this review, we selected several representative viruses that significantly threatened human public health and systematically elucidated the multiple strategies for these viruses or specific viral proteins to antagonize the host antiviral innate immunity, hoping to provide ideas for molecular mechanism research that emerging viruses antagonize the host antiviral innate immunity and accelerate the research progress. Current studies on the viral antagonism of host antiviral innate immunity have focused on the mechanisms of blocking antiviral innate immune signaling pathways. The primary regulatory mechanisms encompass transcriptional regulation, post-translational regulation (such as phosphorylation and K63-linked Ub), preventing complex formation, impeding nuclear translocation, cleavage, and degradation, and so forth. Among these, the autophagy and ubiquitin-proteasome pathways are involved in degradation.
Block antiviral innate immune signaling pathways directly
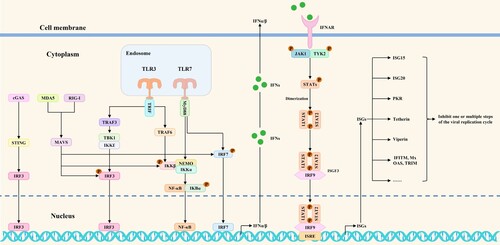
Emerging viral infections may trigger both innate and adaptive immune reactions. Innate immune responses are one of the current research hotspots in immunology. Antiviral innate immune signaling pathways play critical roles in innate immune responses. Pattern recognition receptors (PRRs) (such as cGAS/STING, MDA5, RIG-I, and TLR3/7) could recognize viral nucleic acids and activate transcription factors, including NF-κB, IRF3, and IRF7. During this process, MAVS, MyD88, TRAF3, TRAF6, TRIF, IκBα, TBK1/IKKϵ, and IKKα/IKKβ/NEMO complexes act as essential signaling proteins for signal transduction. These transcription factors cause the production of IFNα/β once they are activated. Following that, IFNα/β binds to interferon receptors and induces the phosphorylation of JAK1, TYK2, STAT1, and STAT2. Phosphorylated STAT1 and STAT2 bind to IRF9 and form ISGF3. The ISGF3 trimers then go to the nucleus, where they bind ISRE to cause the synthesis of ISGs, ultimately preventing the spread of the virus [Citation5]. A schematic diagram of antiviral innate immune signaling pathways mediated by RIG-I, MDA5, TLR3/7, and cGAS/STING is shown in . The battles between viruses and hosts have been constant. Host PRRs recognize viral nucleic acid and then rapidly activate antiviral innate immune signaling pathways to induce ISGs expression, ultimately blocking one or multiple steps of the viral replication cycle. In the meantime, viruses have developed a number of regulatory mechanisms that directly block antiviral innate immune signaling pathways. They could inhibit the expression of interferons and downstream ISGs by targeting and regulating PRRs, signaling proteins, transcription factors, and interferon responses. The initial exploration of these potential mechanisms is an indispensable research direction for emerging virus-host interactions. A summary of these regulatory mechanisms could provide clearer ideas for studying emerging virus-host interactions.
Figure 1. Antiviral innate immune signaling pathways mediated by RIG-I, MDA5, TLR3/7, and cGAS/STING. After virus infection of cells, antiviral innate immune signaling pathways are activated to induce the phosphorylation of transcription factors and IFNα/β expression. Subsequently, IFNα/β binds to interferon receptors on target cells, thereby initiating antiviral interferon responses and, ultimately, ISGs expression.
Target PRRs
As PRRs, cGAS/STING, MDA5, and RIG-I are essential for innate immune responses. Nevertheless, viruses have evolved several strategies to affect how these PRRs work. For example, DENV NS2B targets the cGAS and degrades it in an autophagy-lysosome-dependent mechanism. Such degradation inhibits type I interferon production in the infected cell [Citation12]. It has been demonstrated that SARS-CoV-2 Nsp5 inhibits K63-ubiquitin modification of STING to disrupt the assembly of the STING functional complex and downstream signaling. Furthermore, SARS-CoV-2 ORF3a could bind to STING and block the nuclear accumulation of p65, thereby inhibiting NF-κB signaling [Citation18]. By contrast, DENV(NS2B3) and ZIKV(NS2B3) inhibit type I interferon production in infected cells by cleaving STING. It's interesting to note that the cytoplasmic loop of STING contains the residues R78 and G79, which are crucial factors in the cleavage that ZIKV NS2B3 mediates [Citation15,Citation19].
There are two types of ubiquitylation: degradative (like K48-linked polyubiquitination) and non-degradative (like K63-linked polyubiquitination). K63-linked polyubiquitination of RIG-I is critical for activating antiviral innate immune responses, whereas K48-linked polyubiquitination mediates RIG-I degradation. Several studies have shown that some viruses could rely on their proteins to interact with RIG-I and repress RIG-I-mediated IFN-β production through multiple pathways, such as inhibiting K63-linked polyubiquitination, preventing complex formation, and degradation, etc. These are IAV(NS1), SARS-CoV-2(M, N, and Nsp5), and ZIKV(NS5), among other viruses and viral proteins. Specifically, ZIKV NS5 may specifically bind to RIG-I by interaction with the CARD domain, which would limit K63-linked polyubiquitination of RIG-I, IRF3 activation, and IFN-β production, all of which would inhibit RIG-I signaling [Citation20]. Besides, SARS-CoV-2 M interacts with RIG-I to impede the formation of the RIG-I-MAVS complex and inhibit IRF3 phosphorylation, thereby reducing type I and III interferon production [Citation21]. Additionally, SARS-CoV-2 Nsp5 deprives RIG-I of its ability to activate MAVS by cleaving off the ten most N-terminal amino acids from RIG-I. In other words, Nsp5 cleaves RIG-I after the Q10 residue, preventing RIG-I from interacting with and activating MAVS [Citation22]. Furthermore, through their interactions with RIG-I and inhibition of the RIG-I signaling pathway, IAV(NS1) and SARS-CoV-2(N) similarly repress IFN-β expression [Citation23,Citation24].
Target signaling proteins
As a highly essential signaling protein, MAVS signaling is regulated by various viruses at multiple levels. These regulatory mechanisms encompass preventing complex formation and degradation, etc. For example, DENV NS4A interacts with MAVS to prevent RIG-I from forming complexes with MAVS, inhibiting RIG-I-induced IRF3 activation and IFN-β expression. The results show that the N-terminal CARD-like (CL) domain and the C-terminal transmembrane (TM) domain of MAVS contribute to the formation of the MAVS-NS4A complex [Citation25]. Besides, ZIKV NS4A interacts with MAVS to prevent RLR from binding to MAVS, which suppresses type I interferons generated by RIG-I and MDA5 [Citation17]. By contrast, the SARS-CoV-2 M protein interacts with MAVS to impair MAVS aggregation and its recruitment of downstream IRF3, TBK1, and TRAF3, attenuating the innate antiviral responses [Citation26]. Studies have indicated that IAV PB1 and SARS-CoV-2 ORF10 could suppress the antiviral innate immune responses by degrading MAVS through autophagy pathways, while SARS-CoV-2 Nsp5 and ZIKV NS3 promote MAVS degradation via the ubiquitin-proteosome pathway [Citation22,Citation27–29]. IAV PB1-F2 and PB2 have been reported to interact with MAVS and inhibit MAVS-mediated IFN-β expression. Further research into the particular molecular regulatory mechanisms may be necessary [Citation30,Citation31].
A number of studies indicate that viruses may block signal transduction of other signaling proteins (including TRAF3, TBK1/IKKϵ, and IKKα/IKKβ/NEMO complexes) in addition to MAVS. These viruses may do this by a variety of mechanisms, such as post-translational regulation, preventing complex formation, and degradation, etc. Mechanistically, IAV NS1/126-225 interacts with TRAF3 to reduce K63-linked ubiquitination of TRAF3 and inhibit IRF3 phosphorylation, thereby decreasing IRF3-mediated IFN-β expression [Citation32]. Besides, by impeding K63-linked polyubiquitination and TRAF3 activation, IAV PB2 may counteract the innate antiviral responses [Citation33]. By contrast, MERS-CoV M interacts with TRAF3 and disrupts the TRAF3-TBK1 association, leading to reduced IRF3 activation [Citation7]. As for TBK1/IKKϵ and IKKα/IKKβ/NEMO complexes, viruses also disrupt their signaling functions at various levels. Specifically, SARS-CoV-2(Nsp13) and ZIKV(NS5) could interact with TBK1 to inhibit its phosphorylation and impair the phosphorylation and nuclear translocation of IRF3, thereby reducing type I interferon production [Citation34,Citation35]. In addition, SARS-CoV-2 M may interact with TBK1 and induce its degradation via K48-linked ubiquitination. The reduced TBK1 further impairs the formation of the TRAF3-TANK-TBK1-IKKϵ complex, which leads to the inhibition of type I interferon production [Citation36]. Furthermore, ZIKV NS5 binds to IKKϵ and inhibits its phosphorylation and expression, blocking IRF3 activation and interferon production [Citation37]. Numerous investigations have demonstrated that by interacting with TBK1/IKKε and preventing complex formation, EBOV(VP35), MERS-CoV(ORF4b), and SARS-CoV(M) could inhibit IRF3 phosphorylation and IFN-β expression [Citation9,Citation38,Citation39]. Additionally, the IAV NS1 protein specifically inhibits IKK-mediated NF-κB activation and the production of downstream antiviral genes by physically interacting with IKKα and IKKβ through the C-terminal effector domain [Citation40]. Moreover, IAV PB1-F2 interacts with IKKβ to inhibit NF-κB binding to DNA and NF-κB signaling [Citation41]. It's interesting to note that during viral infection, SARS-CoV-2 ORF9b is known to target the NEMO and disrupt its K63-linked polyubiquitination, which inhibits IKKα/β/γ-NF-κB signaling and subsequent interferon production [Citation42].
Target transcription factors and interferon responses
It is commonly recognized that the transcription factors NF-κB and IRF3 play a crucial role in regulating the expression of IFNα/β that is brought on by viral infections. However, the outcomes demonstrated that IAV, SARS-CoV, and SARS-CoV-2 could regulate the activity and nuclear translocation of IRF3 to inhibit IFNα/β expression. Mechanistically, IAV(PA) and SARS-CoV(PLpro) reduce IFN-β expression by inhibiting IRF3 phosphorylation [Citation43,Citation44]. By contrast, SARS-CoV(8b, 8ab) could target IRF3 and induce its degradation through ubiquitin-proteasome pathways [Citation45]. Moreover, IAV(PA), SARS-CoV(PLpro), and SARS-CoV-2(Nsp5, Nsp12, and ORF3b) could suppress type I interferon production by interacting with IRF3 and preventing its nuclear translocation [Citation43,Citation44,Citation46–48]. Viruses can target and modulate the nuclear translocation of NF-κB in addition to IRF3 in order to disrupt signaling pathways. IAV PA-X specifically inhibits NF-κB p65 nuclear translocation and NF-κB activity, which suppresses IFN-β production [Citation49].
Once activated, transcription factors bind to the interferon promoter to regulate interferon expression and induce interferon responses. The interferon-activated JAK-STAT signaling pathway is essential for ISGs production. Nevertheless, several viruses could block it through a variety of mechanisms. According to reports, ZIKV NS2B3 promotes JAK1 degradation in a proteasome-dependent manner, hence inhibiting the JAK-STAT signaling pathway [Citation50]. Multiple studies have shown that DENV, HIV-1, SARS-CoV-2, and ZIKV could inhibit the signaling of STAT1 and STAT2 to antagonize the host antiviral innate immunity. These antagonistic mechanisms include inhibiting phosphorylation and degradation. Specifically, SARS-CoV-2(Nsp13) interacts with STAT1 to inhibit its phosphorylation, whereas HIV-1(Vif) and SARS-CoV-2(Nsp5) promote its degradation through the ubiquitin-proteasome and autophagy pathways, respectively [Citation6,Citation8,Citation51]. In addition, DENV(NS5) and ZIKV(NS5) suppress interferon signaling by inducing STAT2 degradation via the ubiquitin-proteasome pathways [Citation13,Citation14]. Besides, SARS-CoV-2 N could block interferon responses by suppressing the phosphorylation of STAT1 and STAT2 [Citation52]. By contrast, ZIKV(NS2A) degrades both STAT1 and STAT2 through ubiquitin-proteasome pathways [Citation16]. Studies have indicated that SARS-CoV-2 N, ORF6, and ORF8 inhibit the ISRE activation induced by SeV infection to suppress interferon signaling [Citation52,Citation53]. PKR, an ISG, detects viral RNA in the cytoplasm and phosphorylates eIF2α to inhibit viral protein synthesis and viral replication. However, EBOV(VP35), IAV (NS1), and MERS-CoV(ORF4a) could block PKR activation to antagonize the host antiviral innate immunity. Mechanistically, EBOV VP35 could antagonize PKR activity through its C-terminal interferon inhibitory domain [Citation10]. Furthermore, the results of the experiments demonstrate that PKR activation is inhibited by direct PKR binding to the IAV NS1. The NS1 amino acid sequence (123–127) is necessary to inhibit PKR activation in virus-infected cells [Citation54]. Additionally, MERS-CoV ORF4a suppresses PKR-mediated translation inhibition through its dsRNA-binding domain [Citation55]. The viral strategies to directly block multiple steps of antiviral innate immune signaling pathways are summarized in . This summary might help us better understand the relationships among viruses (viral proteins), regulated targets, and steps of blocked signaling pathways.
Table 1. The viral strategies to directly block multiple steps of antiviral innate immune signaling pathways.
Block antiviral innate immune signaling pathways indirectly by hijacking ncRNAs and proviral host factors or antagonizing host restriction factors
Host ncRNAs are well established to be essential regulators of host biological processes, including apoptosis, inflammation, and viral replication [Citation56]. It has been demonstrated that viruses could also antagonize the host antiviral innate immunity by upregulating the expression of microRNAs, lncRNAs, circRNAs, and vtRNAs to block multiple steps of antiviral innate immune signaling pathways indirectly. “Hijacking” refers to the positive regulatory effects of viruses on the expression of certain host biomolecules.
MicroRNAs are a kind of ncRNAs with approximately 20 nucleotides in length. According to a number of studies, when viruses seize control of microRNAs, they typically attach to the 3′ untranslated regions (3′UTRs) of the target genes to prevent transcription and protein synthesis, which in turn prevents signal transduction. For example, miR-485 suppresses RIG-I expression after viral infection by directly binding to its 3′UTR. Further studies found that the suppression was mediated through posttranscriptional silencing by degrading the RIG-I mRNA. Besides, this interaction substantially reduces the RIG-I-mediated antiviral response, promoting IAV replication [Citation56]. Furthermore, Li et al. discovered that IAV infection could trigger the synthesis of miR-125a, which inhibits the translation of MAVS mRNA by binding to a functional location on its 3′-UTR. Further studies indicated that inhibition of MAVS expression by miR-125a reduces IFN-β responses, leading to increased viral replication [Citation57]. Remarkably, the outcomes of the experiments indicate a considerable rise in miR-146a expression following viral infections. Moreover, overexpression of miR-146a promoted DENV and IAV replication, while downregulation of miR-146a repressed replication. Further investigation revealed that the proviral role of miR-146a was mediated by targeting TRAF6 and inhibiting IFN-β production. These findings suggested that miR-146a may be a potential therapeutic target in viral infections since DENV and IAV could hijack it to target TRAF6 and reduce its expression, inhibiting interferon production [Citation58,Citation59].
Long noncoding RNA (lncRNA) is a class of ncRNA greater than 200 nucleotides in length. Numerous biological processes, including host antiviral responses and IAV replication, are regulated by lncRNAs, according to studies. Besides, it has been found that IAV could hijack lncRNAs to block antiviral innate immune signaling pathways indirectly. Specifically, Jiang et al. discovered that during IAV infection, monocytes had significant levels of lncRNA-NSPL expression. Furthermore, NSPL overexpression makes animals more prone to IAV infection than WT mice, while NSPL knockdown dramatically reduces IAV replication in THP-1 cells. Additional research revealed that by binding to RIG-I and obstructing the interaction between RIG-I and TRIM25, lncRNA-NSPL might impede TRIM25-mediated K63-linked ubiquitination of RIG-I and inhibit antiviral innate immune responses [Citation60]. In addition, Li et al. identified a lncRNA named lncRNA-MxA, which is upregulated after IAV infection. They found that the lncRNA-MxA overexpression facilitates IAV replication, while lncRNA-MxA knockdown inhibits it. The IFN-β promoter may form triplexes with lncRNA-MxA, which would prevent NF-κB and IRF3 from binding to the promoter and hence inhibit the transcription of IFN-β, according to more research. These findings show that IAV may induce lncRNA-MxA expression to inhibit IFN-β transcription and antagonize the host antiviral innate immunity [Citation61]. Additionally, Wang et al. observed that lncRNA-TSPOAP1-AS1 was significantly induced in A549 cells after IAV infection and poly (I:C) stimulation. Interestingly, TSPOAP1-AS1 induction by IAV infection was regulated by the NF-κB signaling pathway. Subsequent research revealed that TSPOAP1-AS1 promotes IAV replication by suppressing ISRE activation and the production of downstream ISGs [Citation62].
Circular RNAs (circRNAs) are a class of ncRNA molecules that lack a 5′ end cap and a 3′ terminal poly(A) tail. Interestingly, Qiu et al. identified one circRNA (circ-MerTK) whose expression was upregulated during IAV infection. The findings showed that, in comparison to the empty vector control during infection, circ-MerTK overexpression reduced the mRNA levels of Mx1, ISG15, and IFN-β. Besides, NIH/3T3 cells expressing sh-circMerTK increased the expression of Mx1, IFITM3, and IFN-β, suggesting that IAV infection could induce circ-MerTK expression to regulate the expression of IFN-β and some ISGs negatively, thereby enhancing viral replication [Citation63]. The vault RNAs (vtRNAs) are a class of 84-141-nt long eukaryotic ncRNAs that RNA polymerase III transcribes. As of right now, vtRNA1-1, vtRNA1-2, vtRNA1-3, and vtRNA2-1 are the four vtRNAs that have been found in human cells. IAV infection induces the host vtRNAs expression through its NS1 protein, as discovered by Li et al. The vtRNAs, hijacked by IAV, enhance viral replication by inhibiting the activation of the antiviral protein PKR and the subsequent interferon expression [Citation64]. The viral strategies to indirectly block antiviral innate immune signaling pathways by hijacking ncRNAs are summarized in .
Figure 2. The viral strategies to indirectly block antiviral innate immune signaling pathways by hijacking ncRNAs and proviral host factors or antagonizing host restriction factors. Red, blue, purple, peacock blue, green, and light blue indicate viral proteins (viruses), microRNAs, lncRNAs, vtRNAs, proviral host factors, and host restriction factors, respectively.
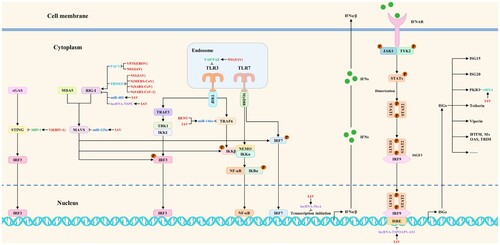
Host factors related to viral replication include proviral host factors and host restriction factors. Proviral host factors directly or indirectly promote viral replication, whereas host restriction factors inhibit it. In addition to upregulating the expression of ncRNAs, viruses could also antagonize the host antiviral innate immunity by hijacking proviral host factors or antagonizing host restriction factors to block multiple steps of antiviral innate immune signaling pathways indirectly. As a proviral host factor, SHP-1 could interact with STING to inhibit K63-linked ubiquitination of STING, thereby suppressing type I interferon production. To dampen the antiviral responses, the association of HIV-1 Vif with SHP-1 facilitates SHP-1 recruitment to STING and inhibits the K63-linked ubiquitination of STING by dephosphorylating STING at Tyr162 [Citation65]. Interestingly, IAV NS1 hijacks YAP/TAZ to suppress the TLR3-mediated innate immune responses. Mechanistically, NS1 could activate YAP/TAZ by direct interaction with YAP/TAZ through their C-terminal TAD. TLR3 expression is downregulated by the activated YAP/TAZ, which inhibits the antiviral innate immune signaling pathways [Citation66]. These studies extend the research direction on the molecular mechanisms by which viruses antagonize the host antiviral innate immunity. The viral strategies to indirectly block antiviral innate immune signaling pathways by hijacking proviral host factors are summarized in .
As a host restriction factor, TRIM25 consists of a SPRY domain, a RING-finger domain, a coiled-coil dimerization domain, and two B-box domains. The SPRY domain interacts with RIG-I CARDs, and the RING-finger domain with E3 ligase activity mediates Lys 63-linked ubiquitination of the N-terminal CARDs of RIG-I, thereby activating the RIG-I pathway and increasing interferon production [Citation67]. Nonetheless, it has been reported that IAV(NS1), MERS-CoV(N), SARS-CoV(N), and SARS-CoV-2(N) could impair the interaction of TRIM25 with RIG-I to inhibit TRIM25-mediated RIG-I ubiquitination and indirectly block antiviral innate immune signaling pathways. Mechanistically, RIG-I signal transduction could be suppressed by IAV NS1 by inhibiting TRIM25-mediated RIG-I CARD ubiquitination. When NS1 interacts with the coiled-coil domain of TRIM25, it blocks TRIM25 multimerization and RIG-I CARD ubiquitination. This connection is mediated by residues E96 and E97 [Citation68]. Additionally, the MERS-CoV N competes with RIG-I for interaction with TRIM25, thereby inhibiting K63-linked ubiquitination of RIG-I CARDs and RIG-I-mediated type I interferon induction [Citation67]. Furthermore, the C terminus of the SARS-CoV N protein interacts with the TRIM25 SPRY domain to inhibit TRIM25-mediated RIG-I ubiquitination and interferon production by preventing the association between TRIM25 and RIG-I [Citation69]. Moreover, SARS-CoV-2 N could also block antiviral innate immune signaling pathways by interacting with the TRIM25 SPRY domain and inhibiting TRIM25-mediated RIG-I activation [Citation70]. PACT is a double-stranded RNA-binding protein that has been shown to interact with the carboxy-terminal domain of RIG-I, promoting RIG-I signaling and host interferon responses. Besides, it could also inhibit viral RNA synthesis and EBOV replication, suggesting that PACT is an essential regulator of RIG-I-mediated signaling pathways and viral replication [Citation11]. Interestingly, research has suggested that EBOV(VP35) and IAV(NS1) may directly interact with PACT to prevent PACT from interacting with and activating RIG-I, therefore counteracting RIG-I-mediated interferon responses [Citation11,Citation71]. The counteracting effects of viruses on host antiviral responses are called “antagonizing”. The viral strategies to indirectly block antiviral innate immune signaling pathways by antagonizing host restriction factors are summarized in . These mechanisms summarized above may provide new perspectives for studying emerging virus-host interactions.
Regulation of metabolic pathways, stress granule formation, apoptosis, and ER stress
In addition to directly or indirectly blocking antiviral innate immune signaling pathways, viruses also antagonize the host antiviral innate immunity by regulating metabolic pathways, the formation of stress granules, apoptosis, and ER stress. These mechanisms might also be one of the research hotspots in emerging virus-host interactions. Specifically, lactate dehydrogenase A (LDHA) expression and LDHA-mediated lactate production were observed to be elevated in response to IAV infection by Thyrsted et al. Interestingly, lactate promotes IAV replication by inhibiting MAVS-dependent induction of type I interferons in primary human airway epithelium. These results indicated that IAV infection could induce lactate formation, which would counteract host antiviral innate defense by reducing interferon production and ISGs expression [Citation72]. Besides, as the omega-3 polyunsaturated fatty acid (PUFA)-derived lipid mediator, protectin D1 (PD1) is reported to reduce IAV replication by inhibiting nuclear export of viral transcripts. Remarkably, during IAV infection, there was a substantial inhibition in PD1 production [Citation73]. Furthermore, Singh et al. showed that AMPK activation has an antiviral effect and inhibits virus-induced glycolysis. In-depth studies revealed that ZIKV could antagonize the host antiviral innate immunity by reducing AMPK activation [Citation74]. One potential way to treat patients with critical diseases might be to target these metabolic alterations. In response to environmental stimuli, including heat shock, oxidative stress, malnutrition, and viral infections, eukaryotic cells can initiate a cascade of reactions. One such response is the formation of stress granules (SGs) to help the cells cope with environmental pressures. Viral infections are one of the conditions for inducing the formation of SGs. The viruses might employ the host translational machinery to complete their life cycles after invading the host cell. Nevertheless, in order to create SGs and fend off viral invasion, the hosts might pause translation. Scientists’ interest in the connection between SGs and antiviral innate immunity has grown during the past few years. During the study, it was surprisingly found that SARS-CoV-2, SARS-CoV, MERS-CoV, and ZIKV could rely on their proteins to inhibit the formation of SGs and promote viral replication [Citation75]. For example, following a coronavirus infection, the viral dsRNA binds to PKR and promotes PKR autophosphorylation, dimerization, and oligomerization. The phosphorylation of eIF2α by the active PKR triggers the recruitment of proteins that nucleate SGs, namely G3BP1, and ultimately results in the assembly of SGs. Nonetheless, the NPs of SARS-CoV-2, SARS-CoV, and MERS-CoV could inhibit PKR phosphorylation and impair SGs formation, promoting a cellular environment propitious for viral replication [Citation75]. Moreover, ZIKV has been shown to inhibit SGs assembly in a phospho-eIF2α-dependent way, which counteracts host antiviral stress responses to enhance viral replication [Citation76].
One of the main mechanisms of programmed cell death connected to the development and etiology of viral diseases is apoptosis. It is essential for viral replication and propagation. Studies have shown that API5, an anti-apoptotic protein, inhibits E2F1-dependent apoptotic signaling and IAV replication. To facilitate E2F1-dependent apoptosis, IAV NP may, nevertheless, repress API5 expression, hence preserving viral replication and dissemination [Citation77]. The endoplasmic reticulum is responsible for protein synthesis, processing, and maturation within the cell. When infecting cells, viruses also utilize the endoplasmic reticulum to complete viral protein synthesis. Simultaneously, the build-up of substantial viral protein quantities triggers a stress response in the endoplasmic reticulum, which subsequently regulates several signaling pathways to preserve cellular homeostasis, leading to the development of autophagy, apoptosis, and metabolic syndrome [Citation78]. Curiously, viruses might have evolved strategies to reduce the inevitable ER stress to a level that is advantageous for viral replication. For example, IAV NS1 interferes with the messenger RNA processing factor CPSF30 and suppresses ER stress response factors, such as XBP1, thereby antagonizing ER stress induction and promoting viral replication [Citation78]. These results provide great ideas for further studying the viral antagonism of host antiviral innate immunity.
Emerging viral strategies to antagonize the host antiviral innate immunity: several future research directions
Research on viral antagonistic mechanisms, as it stands, advances our knowledge of emerging viruses and their pathogenic mechanisms. Therefore, viral strategies to antagonize the host antiviral innate immunity are an indispensable research direction for emerging virus-host interactions. The question of how to investigate the antagonistic mechanisms of emerging viruses more thoroughly and quickly is coming into focus. The most likely solution, without a doubt, is to enhance the learning of research ideas among different viruses. It could provide essential inspiration for studying mechanisms by which emerging viruses antagonize the host antiviral innate immunity. Given this, we try to propose several research directions for SARS-CoV-2, MPXV, and future unknown emerging viruses to antagonize the host antiviral innate immunity based on these summarized viral strategies, which is expected to accelerate the progress of research on emerging viruses to antagonize the host antiviral innate immunity and to deepen the understanding of the pathogenic mechanisms of emerging viruses.
Over the past few years, many studies have been carried out worldwide to investigate the molecular mechanisms by which emerging SARS-CoV-2 antagonizes host antiviral innate immunity. Nevertheless, we find that the molecular mechanism studies that SARS-CoV-2 antagonizes host antiviral innate immunity are often limited to direct regulation of innate immune signaling pathways by viral proteins. Furthermore, the antagonistic mechanisms involving proviral host factors, host restriction factors, ncRNAs (such as lncRNAs, microRNAs, circRNAs, and vtRNAs), metabolic pathways, apoptosis, and ER stress are hardly studied. By contrast, the mechanisms of IAV in these aspects mentioned above are relatively mature, which could provide many important research ideas [Citation56,Citation62–64,Citation66,Citation68,Citation72,Citation77,Citation78]. Additionally, using confocal microscopy, Yi et al. discovered that the PA protein of IAV interacts with IRF3 in the cytoplasm and prevents its phosphorylation, with the N-terminal domain of PA protein playing a crucial role in this process. Further studies revealed that the binding activity of PA protein to IRF3 depends on Asp108, the 108th amino acid in PA protein, and mutation of this aspartate site could weaken the inhibitory effect on IFN-β expression [Citation43]. This discovery provides a substantial reference value for investigating the molecular mechanisms by which SARS-CoV-2 antagonizes host antiviral innate immunity, expanding the research idea of subcellular localization, structural domains, and binding sites of viral-host protein interactions.
MPXV, a member of the Poxviridae family and genus Orthopoxvirus, is the causative agent of monkeypox. MPXV, another representative of emerging viruses, gravely threatens global health security. Worryingly, the current studies on the strategies for MPXV to antagonize the host antiviral innate immunity are still in the initial stages. Some inferences are often drawn from studies performed with the vaccinia virus and related orthopoxviruses [Citation3]. Nevertheless, the lack of a sufficiently structured theoretical underpinning emerging from these research results has caused the MPXV study to proceed slowly. Therefore, the viral strategies to antagonize the host antiviral innate immunity summarized above could also provide new ideas for studying emerging MPXV. For example, from , we discovered that viral strategies directly blocking antiviral innate immune signaling pathways have two crucial features. Multiple viral proteins could work together to antagonize one host factor or signal transduction process. Additionally, one single viral protein may antagonize multiple host factors or signal transduction processes. Given this, we speculated whether some MPXV proteins could synergistically antagonize the same host factor or signal transduction process. Furthermore, screening for MPXV proteins that can antagonize multiple signal transduction processes may be worthwhile. In addition to the connections between viruses (viral proteins), regulated targets, and steps of blocked signaling pathways, the antagonistic mechanisms should not be ignored. In other words, the mechanisms by which viruses or viral proteins regulate their targets mainly include transcriptional regulation, post-translational regulation, preventing complex formation, impeding nuclear translocation, cleavage, and degradation, etc. Which mechanism MPXV employs deserves further study. The current studies on the strategies for MPXV (a DNA virus) to antagonize the host antiviral innate immunity are still in the initial stages. Consequently, the relevant research results of RNA viruses (e.g. IAV and SARS-CoV-2) could provide many new ideas and references for MPXV studies. Borrowing ideas from other viruses (not limited to the vaccinia virus and related orthopoxviruses) could accelerate the progress of the MPXV study.
Undoubtedly, exploring new mechanisms by which emerging viruses antagonize the host antiviral innate immunity is essential because it could help us understand the pathogenic mechanisms of emerging viruses more comprehensively. Based on the mechanisms summarized above, we hypothesize that it may be traditional research ideas to explore how they directly block antiviral innate immune signaling pathways when facing unknown emerging viruses in the future. Consequently, whether emerging viruses could indirectly block antiviral innate immune signaling pathways by hijacking ncRNAs and proviral host factors or antagonizing host restriction factors deserves further study to expand the research direction. Furthermore, whether emerging viruses antagonize the host antiviral innate immunity by regulating metabolic pathways, the formation of stress granules, apoptosis, and ER stress also deserves further exploration. In addition, it is worth pondering which viral proteins regulate each of these processes that antagonize the host antiviral innate immunity and their regulatory mechanisms. In summary, the relatively systematic mechanisms outlined above are expected to accelerate the study's progress on the antagonistic effects of emerging SARS-CoV-2 and MPXV on host antiviral innate immunity, as well as provide a wealth of new research ideas for emerging virus-host interactions. Additionally, it could enable us to more effectively handle future public health threats posed by unknown emerging viruses.
Concluding remarks and future perspectives
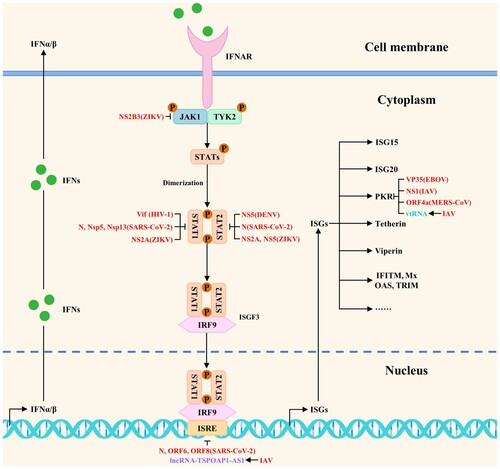
Studies have shown that viruses have evolved multiple strategies to antagonize the host antiviral innate immunity by directly or indirectly blocking the host innate immune signaling pathways during the long game with hosts. The related host proteins include proviral host factors and host restriction factors, whereas the ncRNAs include microRNAs, lncRNAs, circRNAs, and vtRNAs. The viral strategies to block multiple steps of antiviral innate immune signaling pathways are shown in and . In addition, viruses also antagonize the host antiviral innate immunity by regulating metabolic pathways, the formation of stress granules, apoptosis, and ER stress. The current studies on viral strategies to antagonize the host antiviral innate immunity mainly focus on blocking multiple steps of host innate immune signaling pathways, including targeted inhibition of PRRs, signaling proteins, transcription factors, and interferon responses. Besides, we try to propose several research directions for SARS-CoV-2, MPXV, and future unknown emerging viruses to antagonize the host antiviral innate immunity based on these summarized viral strategies.
Figure 3. The viral strategies to block antiviral innate immune signaling pathways: inhibition of interferon induction. Viruses could block interferon production by targeting and regulating multiple steps of antiviral innate immune signaling pathways such as PRRs, signaling proteins, and transcription factors. Red, blue, purple, green, and light blue indicate viral proteins (viruses), microRNAs, lncRNAs, proviral host factors, and host restriction factors, respectively.
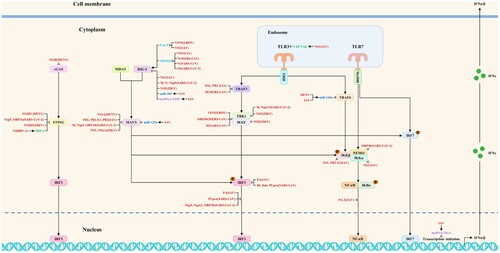
Figure 4. The viral strategies to block antiviral innate immune signaling pathways: inhibition of interferon responses. Viruses could antagonize the host antiviral innate immunity by directly or indirectly blocking interferon responses. Red, purple, and peacock blue indicate viral proteins (viruses), lncRNAs, and vtRNAs, respectively.
Interestingly, viruses can antagonize the host antiviral innate immunity in ways other than the ones listed above. For instance, Timilsina et al. show that SERINC5 could prevent viral fusion during entry, suggesting it is a host restriction factor for SARS-CoV-2. By preventing SERINC5 from being incorporated into progeny virions, SARS-CoV-2 ORF7a can decrease its antiviral function, according to more research. In addition, stimulation with IFN-β does not affect SERINC5 expression levels [Citation79]. These results suggest that SARS-CoV-2 could antagonize a non-ISG host restriction factor, which exerts its antiviral effect by directly targeting the viral replication cycle rather than the antiviral innate immune signaling pathways mentioned above. Besides, studies have shown that IAV NS1 could interact with DNMT3B and promote its K48-linked ubiquitination and degradation. Next, the methylated promoter regions of the JAK-STAT signaling suppressors undergo fast demethylation, thereby increasing their expression and inhibiting JAK-STAT signaling [Citation80]. This study involving epigenetic regulation enriches the understanding of mechanisms by which viruses antagonize the host antiviral innate immunity by indirectly blocking antiviral innate immune signaling pathways. Interestingly, the expression of miR-1249, miR-1307-3p, miR-584-5p, miR-324-5p, and miR-491 is downregulated during viral infections despite the fact that these molecules may directly inhibit IAV replication by targeting and decreasing the expression of viral proteins [Citation5]. These studies further expand the targets of IAV antagonism from host restriction factors to ncRNAs. Beyond that, there may be more new antagonistic mechanisms to be studied and summarized in the future. There needs to be more analysis and discussion of future research trends regarding the viral antagonism of host antiviral innate immunity (Box 1).
Based on previous research results, borrowing research ideas of known antagonistic mechanisms among different viruses, further study of known antagonistic mechanisms, and exploring unknown antagonistic mechanisms may be one of the future research trends regarding the viral antagonism of host antiviral innate immunity. Although current studies on the mechanism by which viruses antagonize the host antiviral innate immunity have formed a relatively systematic theoretical system, there is still a certain gap in the research progress on antagonistic mechanisms among different viruses. For example, there are relatively few research results for EBOV to antagonize the host antiviral innate immunity compared to other representative viruses. Consequently, it is necessary to strengthen the borrowing of research ideas about known antagonistic mechanisms among different viruses. Specifically, more antagonistic mechanisms by which EBOV antagonizes the host antiviral innate immunity by directly blocking antiviral innate immune signaling pathways need to be further expanded. Besides, it remains to be answered whether its antagonistic mechanism is related to proviral host factors, host restriction factors, ncRNAs (such as lncRNAs, microRNAs, circRNAs, and vtRNAs), metabolic pathways, apoptosis, and ER stress. According to the research ideas mentioned above, the antagonistic mechanisms of other viruses could be further explored based on their respective research progress. Furthermore, some studies have not yet elucidated which viral protein(s) mediate viral antagonism of host antiviral innate immunity. They deserve further study, which may deepen the understanding of viral antagonistic mechanisms and provide a theoretical basis for developing antiviral drugs based on these further investigated antagonistic mechanisms in the future [Citation5].
Notably, significant progress has been made in the past few years in elucidating the molecular mechanisms by which different viruses antagonize the host antiviral innate immunity. Nevertheless, there are still several knowledge gaps to be further investigated. Previous studies have shown that host biomolecules associated with viral replication include proteins, ncRNAs, sugars, lipids, hormones, and inorganic salts. In addition to the proteins, ncRNAs, sugars, and lipids mentioned above, it remains to be investigated whether other biomolecules are intrinsically linked to viral antagonism of host antiviral innate immunity [Citation5]. If so, how do viruses regulate them? With the rapid development of single-cell technologies, the mechanisms of virus-host interactions are beginning to be explored at the single-cell level [Citation5]. It is challenging to elucidate the molecular mechanisms by which different viruses antagonize the host antiviral innate immunity at the single-cell level, especially the regulation mechanisms between viruses and host biomolecules (not limited to proteins). Finally, it will be an enormous challenge for future researchers to explore the viral dynamic antagonism of host antiviral innate immunity, especially the dynamic interactions between viral proteins and host biomolecules. These will be interesting topics for future research. Moving forward, we must constantly extend our knowledge of viral strategies to antagonize the host antiviral innate immunity.
The World Health Organization has recently called for preparedness to combat the next wave of widespread infectious diseases brought on by unknown viruses or pathogens and has been researching the likelihood of a devastating pandemic brought on by an unknown disease. Given this, mechanism studies and summarization of strategies for different viruses to antagonize the host antiviral innate immunity are necessary. In other words, there is an urgent need to constantly extend our knowledge of viral strategies to antagonize the host antiviral innate immunity. In this manner, when confronted with the next emerging viral outbreak, we may quickly conduct investigations of the pertinent molecular mechanisms and draw on prior research to obtain a more thorough understanding of emerging viruses and their pathogenic mechanisms. In this manner, we may continue to defend human public health with knowledge and promptly respond to public health threats from unknown emerging viruses in the future.
Glossary
Acknowledgments
The authors thank members of the Ping laboratory for helpful discussions. Na Chen performed the systematic literature review and wrote the manuscript. Jiayu Jin, Baoge Zhang, Qi Meng, Yuanlu Lu, Bing Liang, Lulu Deng, Bingchen Qiao, and Lucheng Zheng revised the manuscript. Jihui Ping organized and provided the frame for the manuscript and critically revised it. All authors read and approved the final manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- Ramasamy S, Subbian S. Critical determinants of cytokine storm and Type I interferon response in COVID-19 pathogenesis. Clin Microbiol Rev. 2021;34(3). doi:10.1128/CMR.00299-20
- Zhang X, et al. SARS-CoV-2 omicron strain exhibits potent capabilities for immune evasion and viral entrance. Signal Transduct Target Ther. 2021;6(1):430. doi:10.1038/s41392-021-00852-5
- Lum FM, et al. Monkeypox: disease epidemiology, host immunity and clinical interventions. Nat Rev Immunol. 2022;22(10):597–613. doi:10.1038/s41577-022-00775-4
- Zhang Q, et al. H7n9 influenza viruses are transmissible in ferrets by respiratory droplet. Science. 2013;341(6144):410–414. doi:10.1126/science.1240532
- Chen N, et al. Virus-host interaction networks as new antiviral drug targets for IAV and SARS-CoV-2. Emerg Microbes Infect. 2022;11(1):1371–1389. doi:10.1080/22221751.2022.2071175
- Wu Y, et al. Main protease of SARS-CoV-2 serves as a bifunctional molecule in restricting type I interferon antiviral signaling. Signal Transduct Target Ther. 2020;5(1):221. doi:10.1038/s41392-020-00332-2
- Lui PY, et al. Middle East respiratory syndrome coronavirus M protein suppresses type I interferon expression through the inhibition of TBK1-dependent phosphorylation of IRF3. Emerg Microbes Infect. 2016;5:e39.
- Gargan S, et al. HIV-1 Promotes the degradation of components of the Type 1 IFN JAK/STAT pathway and blocks anti-viral ISG induction. EBioMedicine. 2018;30:203–216. doi:10.1016/j.ebiom.2018.03.006
- Prins KC, et al. Ebola virus protein VP35 impairs the function of interferon regulatory factor-activating kinases IKKepsilon and TBK-1. J Virol. 2009;83(7):3069–3077. doi:10.1128/JVI.01875-08
- Schumann M, et al. Ebola virus VP35 antagonizes PKR activity through its C-terminal interferon inhibitory domain. J Virol. 2009;83(17):8993–8997. doi:10.1128/JVI.00523-09
- Luthra P, et al. Mutual antagonism between the Ebola virus VP35 protein and the RIG-I activator PACT determines infection outcome. Cell Host Microbe. 2013;14(1):74–84. doi:10.1016/j.chom.2013.06.010
- Aguirre S, et al. Dengue virus NS2B protein targets cGAS for degradation and prevents mitochondrial DNA sensing during infection. Nat Microbiol. 2017;2:17037. doi:10.1038/nmicrobiol.2017.37
- Ashour J, et al. NS5 of dengue virus mediates STAT2 binding and degradation. J Virol. 2009;83(11):5408–5418. doi:10.1128/JVI.02188-08
- Kumar A, et al. Zika virus inhibits type-I interferon production and downstream signaling. EMBO Rep. 2016;17(12):1766–1775. doi:10.15252/embr.201642627
- Ding Q, et al. Species-specific disruption of STING-dependent antiviral cellular defenses by the Zika virus NS2B3 protease. Proc Natl Acad Sci U S A. 2018;115(27):E6310–E6318. doi:10.1073/pnas.1803406115
- Fanunza E, et al. Zika virus NS2A inhibits interferon signaling by degradation of STAT1 and STAT2. Virulence. 2021;12(1):1580–1596. doi:10.1080/21505594.2021.1935613
- Ma J, et al. Zika virus Non-structural protein 4A blocks the RLR-MAVS signaling. Front Microbiol. 2018;9:1350. doi:10.3389/fmicb.2018.01350
- Rui Y, et al. Unique and complementary suppression of cGAS-STING and RNA sensing- triggered innate immune responses by SARS-CoV-2 proteins. Signal Transduct Target Ther. 2021;6(1):123. doi:10.1038/s41392-021-00515-5
- Aguirre S, et al. DENV inhibits type I IFN production in infected cells by cleaving human STING. PLoS Pathog. 2012;8(10):e1002934. doi:10.1371/journal.ppat.1002934
- Li A, et al. NS5 conservative site is required for Zika Virus to restrict the RIG-I signaling. Front Immunol. 2020;11:51. doi:10.3389/fimmu.2020.00051
- Zheng Y, et al. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) membrane (M) protein inhibits type I and III interferon production by targeting RIG-I/MDA-5 signaling. Signal Transduct Target Ther. 2020;5(1):299. doi:10.1038/s41392-020-00438-7
- Liu Y, et al. SARS-CoV-2 Nsp5 demonstrates Two distinct mechanisms targeting RIG-I and MAVS To evade the innate immune response. mBio. 2021;12(5):e0233521.
- Mibayashi M, et al. Inhibition of retinoic acid-inducible gene I-mediated induction of beta interferon by the NS1 protein of influenza A virus. J Virol. 2007;81(2):514–524. doi:10.1128/JVI.01265-06
- Chen K, et al. SARS-CoV-2 nucleocapsid protein interacts with RIG-I and represses RIG-mediated IFN-beta production. Viruses. 2020;13(1). doi:10.3390/v13010047
- He Z, et al. Dengue virus subverts host innate immunity by targeting adaptor protein MAVS. J Virol. 2016;90(16):7219–7230. doi:10.1128/JVI.00221-16
- Fu YZ, et al. SARS-CoV-2 membrane glycoprotein M antagonizes the MAVS-mediated innate antiviral response. Cell Mol Immunol. 2021;18(3):613–620. doi:10.1038/s41423-020-00571-x
- Zeng Y, et al. The PB1 protein of influenza A virus inhibits the innate immune response by targeting MAVS for NBR1-mediated selective autophagic degradation. PLoS Pathog. 2021;17(2):e1009300. doi:10.1371/journal.ppat.1009300
- Li X, et al. SARS-CoV-2 ORF10 suppresses the antiviral innate immune response by degrading MAVS through mitophagy. Cell Mol Immunol. 2022;19(1):67–78. doi:10.1038/s41423-021-00807-4
- Li W, et al. Zika virus circumvents host innate immunity by targeting the adaptor proteins MAVS and MITA. FASEB J. 2019;33(9):9929–9944. doi:10.1096/fj.201900260R
- Varga ZT, et al. The influenza virus protein PB1-F2 inhibits the induction of type I interferon at the level of the MAVS adaptor protein. PLoS Pathog. 2011;7(6):e1002067. doi:10.1371/journal.ppat.1002067
- Graef KM, et al. The PB2 subunit of the influenza virus RNA polymerase affects virulence by interacting with the mitochondrial antiviral signaling protein and inhibiting expression of beta interferon. J Virol. 2010;84(17):8433–8445. doi:10.1128/JVI.00879-10
- Qian W, et al. The C-terminal effector domain of Non-structural protein 1 of influenza A virus blocks IFN-beta production by targeting TNF receptor-associated factor 3. Front Immunol. 2017;8:779. doi:10.3389/fimmu.2017.00779
- Sun N, et al. TRIM35 mediates protection against influenza infection by activating TRAF3 and degrading viral PB2. Protein Cell. 2020;11(12):894–914. doi:10.1007/s13238-020-00734-6
- Vazquez C, et al. SARS-CoV-2 viral proteins NSP1 and NSP13 inhibit interferon activation through distinct mechanisms. PLoS One. 2021;16(6):e0253089. doi:10.1371/journal.pone.0253089
- Lin S, et al. Zika virus NS5 protein antagonizes type I interferon production via blocking TBK1 activation. Virology. 2019;527:180–187. doi:10.1016/j.virol.2018.11.009
- Sui L, et al. SARS-CoV-2 membrane protein inhibits Type I interferon production through ubiquitin-mediated degradation of TBK1. Front Immunol. 2021;12:662989. doi:10.3389/fimmu.2021.662989
- Lundberg R, et al. Zika virus Non-structural protein NS5 inhibits the RIG-I pathway and interferon lambda 1 promoter activation by targeting IKK epsilon. Viruses. 2019;11(11). doi:10.3390/v11111024
- Yang Y, et al. Middle East respiratory syndrome coronavirus ORF4b protein inhibits type I interferon production through both cytoplasmic and nuclear targets. Sci Rep. 2015;5:17554. doi:10.1038/srep17554
- Siu KL, et al. Severe acute respiratory syndrome coronavirus M protein inhibits type I interferon production by impeding the formation of TRAF3.TANK.TBK1/IKKepsilon complex. J Biol Chem. 2009;284(24):16202–16209. doi:10.1074/jbc.M109.008227
- Gao S, et al. Influenza A virus-encoded NS1 virulence factor protein inhibits innate immune response by targeting IKK. Cell Microbiol. 2012;14(12):1849–1866. doi:10.1111/cmi.12005
- Reis AL, McCauley JW. The influenza virus protein PB1-F2 interacts with IKKbeta and modulates NF-kappaB signalling. PLoS One. 2013;8(5):e63852. doi:10.1371/journal.pone.0063852
- Wu J, et al. SARS-CoV-2 ORF9b inhibits RIG-I-MAVS antiviral signaling by interrupting K63-linked ubiquitination of NEMO. Cell Rep. 2021;34(7):108761. doi:10.1016/j.celrep.2021.108761
- Yi C, et al. Influenza A virus PA antagonizes interferon-beta by interacting with interferon regulatory factor 3. Front Immunol. 2017;8:1051. doi:10.3389/fimmu.2017.01051
- Devaraj SG, et al. Regulation of IRF-3-dependent innate immunity by the papain-like protease domain of the severe acute respiratory syndrome coronavirus. J Biol Chem. 2007;282(44):32208–32221. doi:10.1074/jbc.M704870200
- Wong HH, et al. Accessory proteins 8b and 8ab of severe acute respiratory syndrome coronavirus suppress the interferon signaling pathway by mediating ubiquitin-dependent rapid degradation of interferon regulatory factor 3. Virology. 2018;515:165–175. doi:10.1016/j.virol.2017.12.028
- Fung SY, et al. SARS-CoV-2 main protease suppresses type I interferon production by preventing nuclear translocation of phosphorylated IRF3. Int J Biol Sci. 2021;17(6):1547–1554. doi:10.7150/ijbs.59943
- Wang W, et al. SARS-CoV-2 nsp12 attenuates type I interferon production by inhibiting IRF3 nuclear translocation. Cell Mol Immunol. 2021;18(4):945–953. doi:10.1038/s41423-020-00619-y
- Konno Y, et al. SARS-CoV-2 ORF3b Is a potent interferon antagonist whose activity Is increased by a naturally occurring elongation variant. Cell Rep. 2020;32(12):108185. doi:10.1016/j.celrep.2020.108185
- Hu J, et al. PA-X protein of H5N1 avian influenza virus inhibits NF-kappaB activity, a potential mechanism for PA-X counteracting the host innate immune responses. Vet Microbiol. 2020;250:108838. doi:10.1016/j.vetmic.2020.108838
- Wu Y, et al. Zika virus evades interferon-mediated antiviral response through the co-operation of multiple nonstructural proteins in vitro. Cell Discov. 2017;3:17006. doi:10.1038/celldisc.2017.6
- Feng K, et al. Interactome profiling reveals interaction of SARS-CoV-2 NSP13 with host factor STAT1 to suppress interferon signaling. J Mol Cell Biol. 2021;13(10):760–762. doi:10.1093/jmcb/mjab068
- Mu J, et al. SARS-CoV-2 N protein antagonizes type I interferon signaling by suppressing phosphorylation and nuclear translocation of STAT1 and STAT2. Cell Discov. 2020;6:65. doi:10.1038/s41421-020-00208-3
- Li JY, et al. The ORF6, ORF8 and nucleocapsid proteins of SARS-CoV-2 inhibit type I interferon signaling pathway. Virus Res. 2020;286:198074. doi:10.1016/j.virusres.2020.198074
- Min JY, et al. A site on the influenza A virus NS1 protein mediates both inhibition of PKR activation and temporal regulation of viral RNA synthesis. Virology. 2007;363(1):236–243. doi:10.1016/j.virol.2007.01.038
- Rabouw HH, et al. Middle East respiratory coronavirus accessory protein 4a inhibits PKR-mediated antiviral stress responses. PLoS Pathog. 2016;12(10):e1005982. doi:10.1371/journal.ppat.1005982
- Ingle H, et al. The microRNA miR-485 targets host and influenza virus transcripts to regulate antiviral immunity and restrict viral replication. Sci Signal. 2015;8(406):ra126. doi:10.1126/scisignal.aab3183
- Hsu AC, et al. MicroRNA-125a and -b inhibit A20 and MAVS to promote inflammation and impair antiviral response in COPD. JCI Insight. 2017;2(7):e90443. doi:10.1172/jci.insight.90443
- Wu S, et al. miR-146a facilitates replication of dengue virus by dampening interferon induction by targeting TRAF6. J Infect. 2013;67(4):329–341. doi:10.1016/j.jinf.2013.05.003
- Zhang F, et al. Downregulation of miR-146a inhibits influenza A virus replication by enhancing the type I interferon response in vitro and in vivo. Biomed Pharmacother. 2019;111:740–750. doi:10.1016/j.biopha.2018.12.103
- Jiang J, et al. LncNSPL facilitates influenza A viral immune escape by restricting TRIM25-mediated K63-linked RIG-I ubiquitination. iScience. 2022;25(7):104607. doi:10.1016/j.isci.2022.104607
- Li X, et al. Long noncoding RNA Lnc-MxA inhibits beta interferon transcription by forming RNA-DNA triplexes at Its promoter. J Virol. 2019;93(21):e00786-19.
- Wang Q, et al. Long noncoding RNA TSPOAP1 antisense RNA 1 negatively modulates type I IFN signaling to facilitate influenza A virus replication. J Med Virol. 2022;94(2):557–566. doi:10.1002/jmv.25483
- Qiu H, et al. Influenza A virus-induced circRNA circMerTK negatively regulates innate antiviral responses. Microbiol Spectr. 2023;11(2):e0363722.
- Li F, et al. Robust expression of vault RNAs induced by influenza A virus plays a critical role in suppression of PKR-mediated innate immunity. Nucleic Acids Res. 2015;43(21):10321–10337.
- Wang Y, et al. HIV-1 Vif suppresses antiviral immunity by targeting STING. Cell Mol Immunol. 2022;19(1):108–121. doi:10.1038/s41423-021-00802-9
- Zhang Q, et al. Influenza A virus NS1 protein hijacks YAP/TAZ to suppress TLR3-mediated innate immune response. PLoS Pathog. 2022;18(5):e1010505. doi:10.1371/journal.ppat.1010505
- Chang CY, et al. Middle East respiratory syndrome coronavirus nucleocapsid protein suppresses Type I and Type III interferon induction by targeting RIG-I signaling. J Virol. 2020;94(13). doi:10.1128/JVI.00099-20
- Gack MU, et al. Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe. 2009;5(5):439–449. doi:10.1016/j.chom.2009.04.006
- Hu Y, et al. The severe acute respiratory syndrome coronavirus nucleocapsid inhibits Type I interferon production by interfering with TRIM25-mediated RIG-I ubiquitination. J Virol. 2017;91(8):e02143-16.
- Gori Savellini G, et al. SARS-CoV-2 N protein targets TRIM25-mediated RIG-I activation to suppress innate immunity. Viruses. 2021;13(8). doi:10.3390/v13081439
- Tawaratsumida K, et al. Quantitative proteomic analysis of the influenza A virus nonstructural proteins NS1 and NS2 during natural cell infection identifies PACT as an NS1 target protein and antiviral host factor. J Virol. 2014;88(16):9038–9048. doi:10.1128/JVI.00830-14
- Thyrsted J, et al. Influenza A induces lactate formation to inhibit type I IFN in primary human airway epithelium. iScience. 2021;24(11):103300. doi:10.1016/j.isci.2021.103300
- Morita M, et al. The lipid mediator protectin D1 inhibits influenza virus replication and improves severe influenza. Cell. 2013;153(1):112–125. doi:10.1016/j.cell.2013.02.027
- Singh S, et al. AMP-Activated Protein kinase restricts Zika Virus replication in endothelial cells by potentiating innate antiviral responses and inhibiting glycolysis. J Immunol. 2020;204(7):1810–1824. doi:10.4049/jimmunol.1901310
- Zheng ZQ, et al. SARS-CoV-2 nucleocapsid protein impairs stress granule formation to promote viral replication. Cell Discov. 2021;7(1):38. doi:10.1038/s41421-021-00275-0
- Amorim R, et al. Zika virus inhibits eIF2alpha-dependent stress granule assembly. PLoS Negl Trop Dis. 2017;11(7):e0005775. doi:10.1371/journal.pntd.0005775
- Mayank AK, et al. Nucleoprotein of influenza A virus negatively impacts antiapoptotic protein API5 to enhance E2F1-dependent apoptosis and virus replication. Cell Death Dis. 2015;6(12):e2018. doi:10.1038/cddis.2015.360
- Mazel-Sanchez B, et al. Influenza A viruses balance ER stress with host protein synthesis shutoff. Proc Natl Acad Sci U S A. 2021;118(36). doi:10.1073/pnas.2024681118
- Timilsina U, et al. SARS-CoV-2 ORF7a potently inhibits the antiviral effect of the host factor SERINC5. Nat Commun. 2022;13(1):2935. doi:10.1038/s41467-022-30609-9
- Liu S, et al. Epigenetic modification Is regulated by the interaction of influenza A virus nonstructural protein 1 with the De Novo DNA methyltransferase DNMT3B and subsequent transport to the Cytoplasm for K48-linked Polyubiquitination. J Virol. 2019;93(7):e01587-18.