
ABSTRACT
Influenza A viruses (IAV) impose significant respiratory disease burdens in both swine and humans worldwide, with frequent human-to-swine transmission driving viral evolution in pigs and highlighting the risk at the animal-human interface. Therefore, a comprehensive One Health approach (interconnection among human, animal, and environmental health) is needed for IAV prevention, control, and response. Animal influenza genomic surveillance remains limited in many Latin American countries, including Colombia. To address this gap, we genetically characterized 170 swine specimens from Colombia (2011–2017). Whole genome sequencing revealed a predominance of pandemic-like H1N1 lineage, with a minority belonging to H3N2 and H1N2 human seasonal-like lineage and H1N1 early classical swine lineages. Significantly, we have identified reassortant and recombinant viruses (H3N2, H1N1) not previously reported in Colombia. This suggests a broad genotypic viral diversity, likely resulting from reassortment between classical endemic viruses and new introductions established in Colombia's swine population (e.g. the 2009 H1N1 pandemic). Our study highlights the importance of a One Health approach in disease control, particularly in an ecosystem where humans are a main source of IAV to swine populations, and emphasizes the need for continued surveillance and enhanced biosecurity measures. The co-circulation of multiple subtypes in regions with high swine density facilitates viral exchange, underscoring the importance of monitoring viral evolution to inform vaccine selection and public health policies locally and globally.
Introduction
In addition to its detrimental effects on human and pig health, Influenza A viruses (IAV) also exert a significant economic toll due to decreased productivity in animal production, increased healthcare costs, and potential disruptions to various sectors of the economy [Citation1]. Swine influenza (SI), caused by IAV, is a prevalent respiratory disease in pigs. Symptoms include fever, coughing, lethargy, and loss of appetite [Citation2]. While SI typically results in low mortality rates, its high transmission rates significantly affect pig health and growth performance, leading to substantial economic losses [Citation3,Citation4]. In today's world, globalization, climate change, and pandemics are prevalent, highlighting the necessity of a One Health approach for controlling zoonotic pathogens such as IAV. This approach recognizes the interconnection among human, animal, and environmental health [Citation5,Citation6].
Swine influenza viruses (SIV) are genetically diverse and classified into various genotypes based on their genetic makeup. These genotypes result from frequent reassortment events between swine, avian, and human influenza viruses [Citation7]. The primary SIV genotypes include H1N1, H3N2, and H1N2, each with multiple lineages and sub-lineages reflecting their complex evolutionary history. The segmented genome of IAV contributes to extensive viral diversity. The genetic diversity of SIVs poses challenges for surveillance, vaccine development, and biosecurity, necessitating ongoing monitoring and research to manage and control outbreaks effectively. Only three subtypes (H1N1, H1N2, H3N2) are considered endemic in swine [Citation1]. Hemagglutinin (HA) and neuraminidase (NA) are key proteins driving subtype classification and immune recognition [Citation8]. Genetic and antigenic variations in these proteins drive IAV evolution, influenced by animal movement, geography, and interspecies transmission [Citation9]. The diversity of swine IAV is further shaped by antigenic drift and shift (gene reassortment) within populations, as well as global viral dissemination, potentially facilitated by pig movement and trade [Citation1,Citation10,Citation11].
The human-animal interface continually gives rise to new IAV strains with zoonotic and pandemic potential, as evidenced by the 2009 swine-origin pandemic [Citation12–14]. However, not all swine IAV can infect humans, and not all human IAV affect pigs. Although the H1N1 pandemic virus originated from swine, several reverse zoonoses events allowed its establishment in the swine population worldwide [Citation15]. Post-2009 pandemic, H1N1pdm09 emerged as a predominant swine IAV subtype globally [Citation16]. Genomic surveillance is critical in tracking IAV transmission and evolution and informing public health policy writers [Citation17,Citation18]. Understanding swine IAV evolution remains challenging, particularly in developing countries with limited resources like Colombia.
Although the swine industry is a significant contributor to meat production in Colombia, genomic surveillance of IAV in swine has been limited. Unfortunately, despite increased global knowledge of IAV and the evidence of IAV circulation in Colombia over the past decade [Citation19–24], much remains unknown about IAV diversity in swine in this country. Here, we conducted a whole genome characterization and analysis of IAV originating from pigs between 2011 and 2017 in 207 pig farms distributed across Colombia's main pork-producing regions.
Our findings revealed a predominance of the H1N1pdm09 subtype, although a wide genotypic diversity of reassortant viruses, a novel repertoire of distinct genotypes, and strong evidence of recombination between viruses was also observed. Understanding the viral diversity at the population level is crucial for vaccine selection, health interventions to reduce the impact of IAV on swine health and minimize the risk of transmission between pigs and humans.
Materials and methods
A detailed description and references of the materials and methods used in this study are provided in supplementary material (SM). Briefly, we used 170 residual nasal specimens collected from pigs (up to 10 weeks old) between 2011 and 2017 in 207 commercial operations across Colombia. These specimens tested positive for IAV using real-time reverse transcription polymerase chain reaction (RT–PCR), with those having a Ct value ≤35 selected for virus isolation. Viral isolates (n = 112) and all other samples positive for IAV by RT–PCR but negative to virus isolation (n = 58) were used for whole genome sequencing (WGS) using Oxford Nanopore technology (ONT). RNA extraction, genome amplification, and sequencing procedures followed previously reported protocols described in SM.
Swine and human IAV sequences were obtained from public databases (GISAID [Citation25] and NCBI [Citation26]) and inspected for redundancy and completeness. Phylogenetic analysis was conducted using Bayesian evolutionary analysis in BEAST to estimate evolutionary relationships and origins of the sequences obtained in this study. Genotypic diversity across IAV gene segments was evaluated based on phylogenetic relationships with reference sequences. Virus subtypes were determined using the phylogenetic origin of HA and NA genes. Full-length sequence functionality was assessed using the NCBI Flu Annotation web service. Recombination and reassortment events were detected and visualized using the RDP4 programme, followed by Maximum Likelihood Estimation (MLE) analysis for topology changes. Identification of homologous recombination was followed per guidelines by Boni et al. [Citation27].
Data Availability: Nucleotide sequences obtained during this study were deposited in the NCBI GenBank database under accession numbers provided in SM (S4).
Ethics Statement: This study received approval from the Institutional Review Board of the National Swine Producers Association-PorkColombia and the Animal Research Ethics Committee of the University of Antioquia. Specimen collection adhered to international ethical standards and Colombian regulations for animal research.
Results
RT–PCR testing and virus isolation
Eighty-seven percent out of the selected repository samples (149/170, 87.6%) yielded Ct values ≤35 after confirmatory RT–PCR testing, and 112 swine IAV were successfully isolated. After WGS, complete genomes were obtained from all IAV isolates, while partial genomes or unsuccessful sequencing were derived from the other specimens. Additional data concerning sequencing success, location, year of collection, and RT–PCR test results from the specimens used in this study are presented in the SM (S1).
Influenza A diversity in swine
The overall genetic diversity of swine IAV observed during this study period is illustrated in . Four main viral groups (G1-G4) were identified according to the complete IAV genomes obtained. Virus with a genetic constellation derived from the 2009 pandemic IAV (pdm) being the most frequent virus found (G1, n = 103). However, one IAV H1N1 derived from early classical swine viruses (CS) was also found (G2, n = 1). This classical H1N1 virus had mixed or reassorted genes at the PA, PB1, and PB2 level (). Additionally, seven H1N2 viruses were sequenced (G3) with a single reassorted IAV constellation containing a pandemic backbone and a human seasonal N2. The H3N2 subtype was less frequently observed (G3, G5 and possibly G6). The complete genome obtained for an H3N2 virus had human seasonal (HS) and pandemic segments at the PA, PB1, and PB2 level. Twenty additional viral groups (G5-G24) were classified from the partial genomes obtained, however they may belong to any of the main viral groups identified (G1-G4). In overall, we identified at least seven viral groups representing reassortant viruses containing one or more genes derived from HS or CS while maintaining a pdm backbone (). shows the distribution and characteristics of the viral groups and subtypes observed in this study.
Figure 1. Classification of swine IAV into viral groups and subtypes according to their genetic constellation. Viral groups were obtained according to the genetic constellations of the gene segments and their combinations. Phylogenetic relationships were assessed by Maximum likelihood inference. The frequency of IAV viral group is shown within brackets. Complete genomes were only obtained for viral groups 1–4 (G1-G4). Incomplete genomes are illustrated as different viral group to avoid overestimation of any viral group. Blue: Pandemic origin; Yellow: Early classical swine origin; Purple: Human seasonal origin. Grey: Sequence information not obtained. First row indicates IAV gene segments HA: Hemagglutinin, NA: Neuraminidase, M: Matrix, NP: Nucleoprotein, NS: non-structural protein, PA: polymerase acidic, PB1: polymerase basic 1, PB2: polymerase basic 2.

Table 1. Summary of the genetic constellations and subtypes of the swine influenza viruses originated from Colombia.
Phylogenetic and recombination analysis
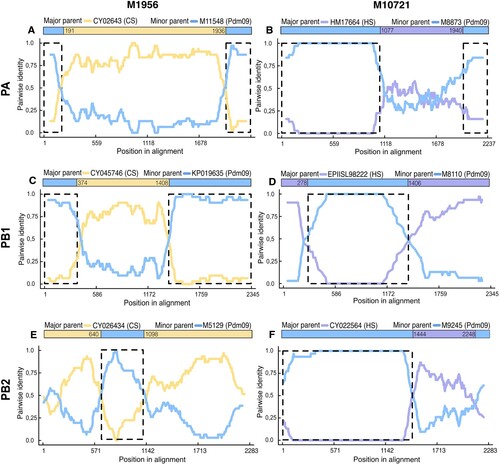
Phylogenetic analysis of PA, PB1, and PB2 genes of two IAV isolates (M1956 and M10721) showed incongruent relationships. Therefore, to determine the involvement of recombinant events in the evolution of these swine IAV, we performed recombinant detection using RDP4 software based on comparing these sequences with the representative IAV strains from different clusters in the phylogenetic tree. Notably, for these two viruses belonging to H1N1 and H3N2 subtypes (from viral groups G2 and G3) the polymerases were recombinant. These suggested recombination events were detected by several of the evaluation methods, and four of them (RDP, GENECONV, BootScan, and MaxChi) were common results for all segments evaluated. The results showed that putatively major recombinant breakpoints in the genome of polymerase genes of these two viruses were detected with a high degree of reliability. The three polymerases of the H1N1 virus-M1956 underwent genetic recombination (at nt positions 191–1936; 374–1408, and 640–1098, respectively) with the major parent derived from early classical swine viruses, while the minor parent derived from H1N1pdm09 viruses. On the other hand, the three polymerases of the H3N2 virus-M10721 underwent genetic recombination (at the nt positions 1940–2286, 279–1406, and 1444–2248, respectively) with the major parent derived from human seasonal viruses while the minor parent derived from 2009 pandemic viruses (see ; and SM ST5). Furthermore, the evolutionary relationships of these recombinant polymerase segments and their putative parental strains were verified by phylogenetic methods as shown in SM. The time-scaled Bayesian approach indicated that the most recent putative ancestor for the H1N1-M1956 recombinant PB1 parental segment circulated in Colombia at least since 2014, while the PB2 parental segment circulated since 2002 (SM figures 8S–10S). Similarly, for the H3N2-M10721 recombinant virus, the PB1 and PBS parental segments have circulated since 2016 (SM figures 11S–13S).
Figure 2. Recombination analysis of polymerases genes (PA, PB1 and PB2) of reassortant viruses from Colombia. Nucleotide pairwise identity (NPI) analysis was conducted using RDP4 programme using the RDP method. Other seven methods were also applied. Each chart represents the NPI between the sequences. The NPI is calculated as the percentage of nucleotides that are identical between the compared sequences, with values ranging from 0% (no identity) to 100% (complete identity). Left panel shows the results for H1N1-M1956, while the right panel shows results for H3N2-M10721. Analysis of the PA gene (A and B); PB1gene (C and D); and PB2 gene (E and F). The nucleotides positions of each recombined region are shown on X axis. Coloured bar at the top of each chart shows the recombined region and the parental virus strain. Detailed information of this analysis and results from the other recombination methods used are shown in SM (S15-22).
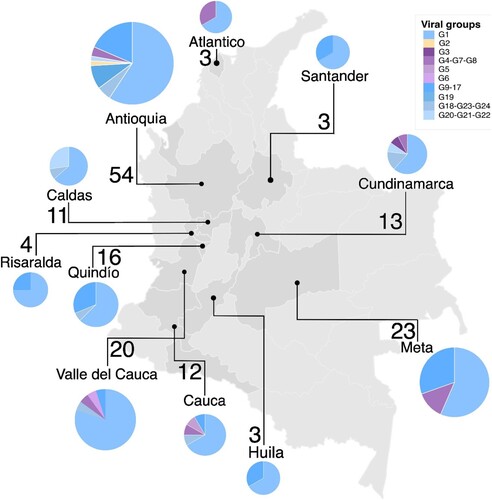
Moreover, as illustrated in , the co-circulation of different genetic constellations and reassortant viruses was observed in several regions (Antioquia, Atlántico, Cauca, Cundinamarca, Meta, and Valle del Cauca) at different time periods (2016 and 2017). Regarding the genetic constellations of internal genes (M, NP, PA, NS, PB1, PB2), many viruses in this study were derived from pdm virus, indicating that most of them acquired the genetic backbone from the H1N1pdm09 virus. Additional details on the virus subtype, genetic constellations, and phylogenetic origin of each gene segment for each virus studied are provided in the SM.
Figure 3. Frequency and spatial distribution of the swine influenza virus in Colombia. Pie charts indicate frequency of viral groups per location. Pie chart colours represent the viral groups according to the genetic constellations of IAV genomes. Values close to the pie charts are number of viruses analysed from that location.
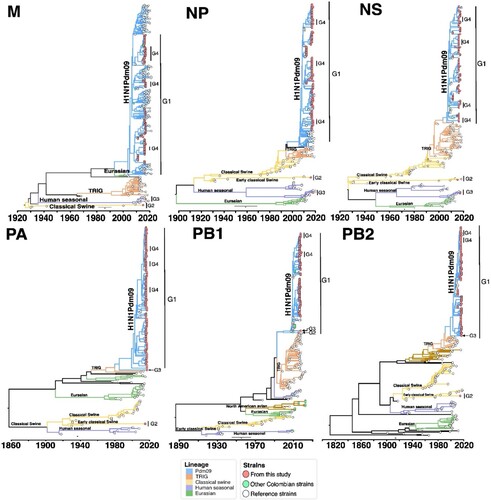
The Bayesian phylogenetic analysis of the IAV genomes from this study revealed insightful patterns regarding the evolutionary dynamics of IAV in Colombia, as illustrated in and . At the HA level, we observed that the majority of H1 viruses were closely related to the pdm lineage, with the exception of one virus (M1956-H1N1) clustering together with early CS viruses. We observed a lower proportion of H3 viruses that clustered into two different clades within the HS lineage. No other lineages of H1 or H3 were identified in this study. Additionally, at the NA gene level, the majority of viruses sequenced were N1 viruses and clustered with pdm viruses or early CS. As the H3 gene was sequenced, the N2 genes clustered into two different clades within the HS lineage. Regarding the phylogenetic relationships of internal genes (M, NP, PA, NS, PB1, PB2), most viruses derived from the pdm lineage, except for two viruses, which derived from early CS and HS lineages. In addition, phylogenetic analyses of the HA, NA, M, NP, and NS gene segments of the M1956-H1N1 recombinant virus revealed clustering with early classical swine viruses reported from China in 2009. Detailed Bayesian phylogenetic trees for each gene segment are presented in the SM.
Figure 4. Bayesian phylogeny of HA (H1 and H3) and NA (N1 and N2) genes of swine IAV in Colombia. Phylogenetic relationships inferred using Bayesian analysis. Each branch is coloured according to the ancestral lineage. Trees were constructed using Beast programme by Bayesian Markov-Chain Monte Carlo with >100,000,000 generations. Red circles represent sequences from this study. Collapsed trees are illustrated here, an extended and more detailed versions are available in supplementary material.
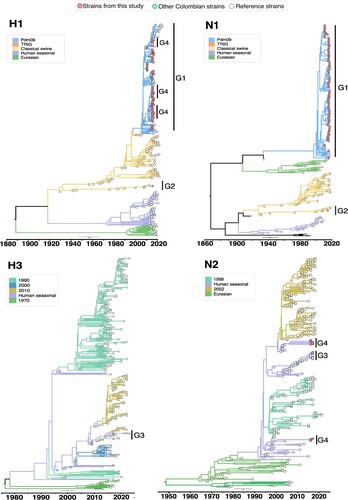
Figure 5. Bayesian phylogeny of internal genes of swine IAV in Colombia. Phylogenetic relationships inferred using Bayesian analysis. Each branch is coloured according to the ancestral lineage. Trees were constructed using Beast programme by Bayesian Markov-Chain Monte Carlo with >100,000,000 generations. Sequences from this study are represented with red circles. Collapsed trees are illustrated here, an extended and more detailed versions are available in supplementary material.
HA protein analysis for H3N2 viruses
The antigenic motif diversity in the HA segment of the two H3N2 viruses from this study was evaluated by comparing the key amino acids at specific positions (145, 155, 156, 158, 159, 189, and 193) against reference antigenic phenotypes [Citation28], the closest phylogenetic human vaccine strain and the current human vaccine strain as shown in . Although antigenic motifs in the M10721 recombinant virus were similar at positions 158 and 159, most of the other motifs greatly differed from the vaccine strains and the other reference antigenic phenotypes. Moreover, the antigenic motif in the M12966 reassortant virus was similar at position 159, but most of the other motifs also differed from the vaccine strains and the other reference antigenic phenotypes.
Table 2. Comparison of antigenic motifs and antigenic phenotypes of H3 Influenza A viruses originated from Colombia.
Discussion
This study provides a comprehensive genome characterization of 162 swine IAV collected from Colombian pig farms between 2011 and 2017. Our findings demonstrate the diversity of swine IAV in Colombia, highlighting the dominance of H1N1 with 2009 pandemic origins, co-circulating with classical H1N1, H1N2, and H3N2 subtypes. Additionally, we evidence reassortment events at multiple gene levels, as potential recombination within the polymerase genes, confirming the complex evolutionary dynamics of IAV in swine. Phylogenetic analysis identified distinct lineages, including pandemic, classical swine, and human-like origin viruses, indicating ongoing genetic divergence. The co-circulation of multiple subtypes in high-density regions likely facilitated viral exchange and reassortment, making IAV harder to control in swine populations and posing a public health risk given the zoonotic potential of IAV. Additionally, the detection of reassortant viruses underscores the need for enhanced surveillance and more robust biosecurity measures to prevent the dissemination of swine IAV or the introduction of human IAV into swine populations.
Consistent with other studies in swine, we observed the three most common IAV swine subtypes (H1N1, H3N2, and H1N2), which are endemic in pigs worldwide [Citation1]. Phylogenetic findings from this study align with similar results reported elsewhere for human and swine IAV circulating in the Americas [Citation29,Citation30]. In agreement to our findings, a study conducted during 2011–2020 found that H1N1, H1N2, and H3N2 viruses have also been circulating in Brazilian swine herds, and that reassortment events were also occurring with H1N1pdm09 viruses [Citation30]. Previous studies have documented the detection and partial characterization of swine IAV in Colombia. Initial reports of H1N1 and H3N2 subtypes date back to the 1970s [Citation31], including several classical swine viruses. Decades later, the 2009 pandemic H1N1 virus, as well as H1N1 viruses from different origins, were reported in swine [Citation20–24]. However, few of these studies investigated the whole genome of these viruses. In this context, our study found that most of the IAV genomes sequenced derived from 2009 pandemic viruses, suggesting a higher fitness of these genes at the viral to persist in swine. However, some viruses still contained genes derived from classical swine and/or human seasonal viruses. Reassortant swine IAV-containing genes from the pandemic virus have also been documented in other world areas [Citation32,Citation33]. Notably, in our study, we observed evidence of recombination in the polymerase genes (PA, PB1, PB2) of two swine IAV (H3N2 and H1N1 subtypes), which may have important implications for viral pathogenesis and transmission; however, further analysis is needed to understand its implications better.
The co-circulation of H1N1pdm09, H3N2, and H1N2 subtypes in the same areas may have facilitated the exchange of viral segments. In our study, H3N2 and H1N2 viruses were clustered with human-like viruses but exhibited a combination of surface genes and/or internal genes (backbone) from H1N1pdm09. These findings suggest that these viruses were introduced from human populations and have been circulating and evolving in the pig populations in Colombia since about a decade. The estimated time of introduction of the HA segment was dated 2002, 2014, and 2015 for viral groups G3, G4, and G5, respectively. While the NA segment was 2016, 2000, and 2006 2015 for viral groups G3, G4, and G5, respectively. The estimated time of introduction for the recombinant polymerase segments was dated around 2012 for M1956 H1N1, while for M10721 H3N2 was around 2015. Interestingly, the co-circulation of H1N1, H1N2, and H3N2 subtypes of IAV was observed in at least three regions during two different years of this study. Such co-circulation of different IAV subtypes has been associated with an increased risk of reassortment events and the emergence of novel IAV viruses with unknown implications for viral pathogenesis and transmission [Citation32,Citation33].
The complete genome of the M1956 H1N1 was phylogenetically related to an early classical swine (CS) virus reported from Iowa (US) in 1930, representing a historical swine H1 IAV [Citation10]. Additionally, the HA and NA genes from this virus clustered together with a CS virus found in Colombia in 2008 [Citation20]. Other genes from M1956 except the polymerases (PA, PB1, and PB2) clustered with an early CS virus from 2009 in China [Citation34]. Due to the lack of genomic data on swine IAV in Colombia, limited conclusions about the origin of the M1956 virus were made and demonstrating the need for increased surveillance of swine IAV in Colombia. However, this virus exhibited close relationships with a virus originating from a designated region considered a putative influenza epicentre [Citation35]. Additionally, there is indirect evidence of the circulation of M1956-related viruses through antibody sero-reactivity against the Iowa 1930 CS strain reported in the Antioquia region of Colombia about half a century ago [Citation36].
The identification of the reassortant and recombinant swine IAVs in this study supports the hypothesis that virus diversity could have resulted from mixed infections within swine herds at production sites or during the transportation of animals between operation sites. However, the involvement of humans in this virus transmission dynamic remains unclear. Mutation (antigenic drift) and dynamic reassortment of genes are considered key components of IAV diversity and evolution [Citation37], and are also related to virulence and changes in host tropism [Citation38]. Avoiding the transmission of IAV between swine populations or from other species to swine is crucial to minimize the impact of IAV on swine and reduce the public health risk. Studies have shown significant evidence demonstrating that homologous recombination occurs between human and swine IAV lineages [Citation39]. Pigs may act as mixing vessels, but humans can also contribute to reassortment during mixed infections, which has a greater impact on health. In 1980, a recombinant strain resembling the 1950 H1N1 IAV reappeared, causing global epidemics alongside the prevailing IAV H3N2 viruses [Citation40]. Also, an exhaustive study in 2008 identified that about 2% of the IAV H3N2 and H1N1 sequences reported in public databases had statistically significant signals of homologous recombination in different RNA segments [Citation41]. Moreover, swine movement during husbandry, trade, or marketing may increase virus cross-infection rates and provide opportunities for virus reassortment [Citation42]. Although the majority of swine in Colombia is raised for domestic consumption, import and export markets involving animal movement from or to neighbouring countries may occur. In addition, pork production in Colombia has grown significantly, driven by a 200% surge in per capita consumption between 2010 and 2023. National production increased by 8.3% from the first quarter of 2023 to the first quarter of 2024, showcasing the industry's ability to adapt and meet evolving market demands [Citation43]. This may have implications for the spread of IAV among swine populations and potentially to humans. As the swine population grows to meet rising demand, there is a greater likelihood of disease transmission among pigs. Several studies have shown that higher densities of pig farms and increased animal movement and transportation within different regions of the country, from and between swine farms, slaughterhouses, and markets, may facilitate the spread of SIV [Citation21,Citation42,Citation44]. Moreover, these increased interactions between humans and pigs can lead to zoonotic or reverse zoonotic transmission of IAVs as demonstrated in other countries [Citation45]. Therefore, effective biosecurity measures and robust monitoring are essential to mitigate the risks associated with SIV in expanding pork production. Most of the swine viruses sequenced in this study, except for one virus, had genetic origins from human strains, highlighting again the important role that humans are playing in the diversity of IAV in pigs in Colombia. Even though pigs have been proposed as a mixing vessel, humans can also be the source of IAV reassortment during mixed infections. The transmission of human IAV to swine populations has been demonstrated in several studies [Citation46].
The swine IAV from this study revealed a wide genotypic diversity in the study period, with most of the viruses belonging to 2009 pandemic lineage (H1N1pdm09). This is consistent with other reports in swine in the region and globally [Citation7,Citation19,Citation29]. The genetic makeup of the pandemic H1 genes analysed in this study and their close relationship with pandemic and seasonal human IAV may suggest that these viruses continuously circulate within the swine population, possibly as a result of multiple reverse zoonotic events as reported in other studies [Citation47–49]. Indeed, the frequent transmission/introduction of human viruses to pigs has resulted in several human-origin virus lineages in swine [Citation11,Citation49]. Transmission of H1N1pdm09 IAV from humans back to the pig population caused the emergence of multiple reassortant genotypes, many of which are now well-established worldwide [Citation11,Citation50]. Viruses containing different constellations of genes derived from H1N1pdm09 are now predominant among IAV circulating in swine in several regions [Citation32,Citation51–53], as seen in Colombia. Adaptations or mutations in viruses of human origin that occurred after transmission to pigs have been reported as a key contribution to the fitness of the IAVs [Citation47]. Also, the persistence of H1N1pdm09 in pigs has been potentially associated with the emergence of novel virus strains with higher transmissibility or pathogenicity in humans and pigs [Citation48,Citation54]. Reassortment of the H1N1pdm09 matrix gene has been linked to increased virus transmission efficiency [Citation55]. Thus, detecting reassortant viruses (H1N1, H1N2, and H3N2) containing genes from the H1N1pdm09 in Colombian pigs raises public health concerns. The H3N2 and H1N2 reassortant viruses have not been previously reported in Colombia, likely due to limited genomic surveillance in the country. Genetic reassorting events of swine IAV have been observed in Argentina, Chile, and Brazil [Citation33,Citation56,Citation57]. Since most of the IAV reported in this study contained backbone (internal genes) from H1N1pdm09, it could be speculated that this condition may have provided stability to the viruses over time, persisting and becoming a predominant lineage in swine during the study period in Colombia. Other authors have also suggested this hypothesis [Citation42].
Previous studies identified seven key amino acid positions (145, 155, 156, 158, 159, 189, 193; H3 numbering throughout) in the HA protein responsible for antigenic drift in human H3 IAV. Some of these were also significant in the antigenic evolution of H3 IAV. Antigenic cartography identified distinct groupings of viruses, known as antigenic clusters, which were differentiated by colour for easy visualization. Antigenic cartography was associated with specific amino acid combinations at these positions, termed “antigenic motifs” [Citation28]. For instance, the Cyan cluster included motifs like NHNNYR, while the Red cluster included motifs like NYNNYK. We did not observe specific trends in antigenic cluster patterns of H3 viruses from this study, and the antigenic motif of these H3 viruses greatly differed from the reference phenotypes currently described [Citation28]. The previously uncharacterized antigenic motifs may represent additional antigenic diversity. However, with this finding alone, we cannot conclude on the antigenic properties of these H3 viruses and their implications. Antigenic mismatch between circulating and vaccine strains may result in significantly reduced effectiveness of vaccines [Citation58]. Similar studies have been conducted for H1 swine IAV viruses from Colombia, describing a high antigenic diversity of these viruses in pigs [Citation19]. Therefore, further studies must be conducted to characterize the antigenic cartography and properties of these H3 viruses circulating in Colombian swine.
Vaccination pressure is known to influence swine IAV epidemiological behaviour. However, in Colombia, where routine swine influenza vaccination is not practiced, IAV can establish causing persistent infections without facing significant evolutionary immune pressure, unlike situations observed in countries where pig vaccination against influenza is common or under experimental conditions [Citation59]. Minor amino acid changes within the antigenic motif of H3 viruses may significantly alter current IAV vaccine efficacy in humans and swine [Citation28]. The diversity in the antigenic motif of the H3 viruses in Colombia should be closely monitored for appropriate vaccine selection, benefiting both swine and humans.
Recent outbreaks of highly pathogenic avian influenza in backyard poultry and wild birds have been reported in Colombia [Citation60]. Consequently, avian, human, and swine IAV are co-circulating in this country. As evidenced, reassortant events between human and swine viruses are occurring, making this scenario ideal for a One Health approach as an integrated strategy for controlling and managing infectious influenza in the country. This approach is crucial for preparing and responding to IAV threats at the human-animal interface [Citation5,Citation61]. Numerous instances of applying this integrated approach have been documented for influenza management [Citation5,Citation62–67], and such examples are worth examining for potential adaptation to the Colombian context.
Implementing enhanced surveillance and biosecurity for swine IAV in Colombia and other Latin American countries faces significant challenges. Major barriers include limited resources and funding, constrained public health budgets, and inadequate infrastructure [Citation68]. To overcome this, international financial support and public-private (e.g. industry-universities) partnerships are essential solutions. The lack of diagnostic capacities, adequate laboratory facilities, diagnostic tools, and data management systems also hampers effective surveillance and response efforts. There is a need for more funding support for animal health surveillance and zoonotic-pandemic preparedness in these regions. Addressing this issue requires significant investment in local infrastructure development, effective biosecurity plans, improved coordinated surveillance systems, and robust training programmes for laboratory and field personnel [Citation69]. Enhancing local capabilities can also be supported by establishing mobile diagnostic units to support areas where the regional reference laboratories cannot provide necessary diagnostic support and expertise. Additionally, the shortage of trained personnel and cultural resistance also complicate efforts. Capacity-building initiatives, including training and education programmes for local professionals and community engagement campaigns, are critical to developing the necessary expertise and fostering trust. Partnerships with universities and research institutions can facilitate continuous education and knowledge sharing, ensuring local personnel are well-equipped to handle surveillance and biosecurity tasks. Community engagement and education campaigns are essential to raise awareness about the importance of swine influenza surveillance and biosecurity. Finally, regulatory challenges, including inconsistent or inadequate regulatory frameworks for animal health, can impede coordinated surveillance efforts. Therefore, harmonizing regulatory frameworks across countries with international standards can improve coordinated surveillance efforts [Citation69].
The COVID-19 pandemic has profoundly impacted various aspects of public health, and scarce information is available on how this may have affected the swine industry. Our study was conducted before the COVID-19 pandemic; therefore, the current SI dynamics scenario may differ. Changes in biosecurity measures (homestay of workers presenting flu-like symptoms) were applied in swine farms during and after the pandemic. Also, reduced movement of people and goods likely occurred during the pandemic. Fewer workers and visitors accessing swine farms could have decreased the risk of introducing and spreading IAV. Disruptions in the supply chain and changes in pork market demands during the pandemic may have also affected the regular practices in swine management, potentially leading to stress and health issues in swine populations that could influence disease dynamics. Also, farms and swine production facilities likely implemented stricter biosecurity measures to prevent the introduction of the disease. Restrictions on the flux of people and goods during the pandemic may have led to reduced movement of swine, potentially limiting the spread of IAV between regions and countries. Therefore, new studies are required to better understand the current dynamics of influenza among both swine and humans in Colombia after COVID-19.
We recognize that this study has certain limitations. The specimens represented a cross-sectional specific period and a subset of the country's pork producers, so they may not fully reflect current geographical or spatiotemporal patterns of IAV circulation in the country. However, while our results contribute to filling the gap in genomic surveillance of IAV, they also provide regional knowledge regarding IAV in pigs.
Conclusions
This study presents a thorough genetic analysis of swine influenza A viruses (IAV) obtained from Colombian pig farms from 2011 to 2017. It delineates the prevalence of the H1N1pdm09 virus within swine populations alongside other subtypes, including H1N1, H1N2, and H3N2. Recombination and reassignment events in viral genes suggest intricate evolutionary dynamics. Phylogenetic analysis identifies distinct lineages, encompassing both pandemic and human-like origin viruses, with evidence of ongoing genetic divergence. The co-circulation of multiple subtypes in regions with high pig density facilitates viral exchange, posing significant public health risks. Moreover, this study suggests the potential involvement of humans in the reassortment of viruses. It underscores the necessity for enhanced surveillance and implementation of robust biosecurity measures. Continued genomic surveillance is essential for vigilantly monitoring viral evolution and informing effective control strategies.
Furthermore, the findings of this study highlight the importance of adopting a comprehensive One Health approach to combat the spread of influenza viruses effectively. Human, animal, and environmental health interconnectedness necessitates a holistic strategy that integrates insights from multiple disciplines. By recognizing the interdependencies between human and animal health and the environment, the One Health approach aims to mitigate the risks posed by infectious diseases like swine influenza. This entails collaborative efforts among public health professionals, veterinarians, environmental scientists, policymakers, and other stakeholders to address the complex factors influencing disease transmission and emergence.
Author contributions
Concept and design: KC, JU; Acquisition, analysis, or interpretation of data: All authors; Drafting of the manuscript: All authors. Critical revision of the manuscript for important intellectual content: All authors. Data analysis: KC, JU. Experiments design, samples processing, data analysis: KC, LP, IM, JU; Obtained funding: KC, JO, JPHO; Administrative, technical, or material support: JO, JPHOs; Supervision: KC, JO, JPHO.
Supplemental Material
Download MS Excel (64.9 KB)Supplemental Material
Download MS Word (2.5 MB)Supplemental Material
Download PDF (38.6 MB)Acknowledgements
We would like to acknowledge the work of all collaborating staff, including veterinarians and laboratory personnel. We thank Simon Villegas, Elizabeth Misas, Maria Stella Lopez, Manuela Rodriguez, and Sebastian Diaz for their assistance.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Anderson TK, Chang J, Arendsee ZW, et al. Swine influenza a viruses and the tangled relationship with humans. Cold Spring Harb Perspect Med [Internet]. 2021;11:1–25. Available from: https://pubmed.ncbi.nlm.nih.gov/31988203/.
- Ma W. Swine influenza virus: current status and challenge. Virus Res [Internet]. 2020;288:198118. Available from: https://linkinghub.elsevier.com/retrieve/pii/S016817022031025X.
- Deblanc C, Simon G. Involvement of swine influenza A viruses in the porcine respiratory disease complex. Virologie [Internet]. 2017;21:225–238. Available from: doi:10.1684/vir.2017.0709.
- Kasowski EJ, Garten RJ, Bridges CB. Influenza pandemic epidemiologic and virologic diversity: reminding ourselves of the possibilities. Clin Infect Dis [Internet]. 2011;52:S44–S49. Available from: doi:10.1093/cid/ciq010.
- Kibenge FSB. A One Health approach to mitigate the impact of influenza A virus (IAV) reverse zoonosis is by vaccinating humans and susceptible farmed and pet animals. Am J Vet Res [Internet]. 2023;84:1–9. Available from: doi:10.2460/ajvr.23.03.0053.
- Kahn RE, Ma W, Richt JA. Swine and influenza: a challenge to one health RESEARCH. 2014. p. 205–218. Available from: doi:10.1007/82_2014_392.
- Rabalski L, Kosinski M, Cybulski P, et al. Genetic diversity of Type A influenza viruses found in swine herds in northwestern Poland from 2017 to 2019: the one health perspective. Viruses [Internet]. 2023;15:1893. Available from: doi:10.3390/v15091893.
- Janke BH. Influenza A virus infections in swine. Vet Pathol [Internet]. 2014;51:410–426. Available from: https://pubmed.ncbi.nlm.nih.gov/24363301/.
- Powell JD, Abente EJ, Chang J, et al. Characterization of contemporary 2010.1 H3N2 swine influenza A viruses circulating in United States pigs. Virology [Internet]. 2021;553:94–101. Available from: doi:10.1016/j.virol.2020.11.006.
- Rajao DS, Anderson TK, Kitikoon P, et al. Antigenic and genetic evolution of contemporary swine H1 influenza viruses in the United States. Virology [Internet]. 2018;518:45–54. Available from: doi:10.1016/j.virol.2018.02.006.
- Lewis NS, Russell CA, Langat P, et al. The global antigenic diversity of swine influenza A viruses. Elife [Internet]. 2016;5:e12217. Available from: doi:10.7554/eLife.12217.
- Yan HX, Xu HF, He WJ, et al. Phylogenetic analysis of HA and NA genes of influenza H1N1 viruses from 1918 to 2017. Acta Virol [Internet]. 2019;63:195–202. Available from: http://www.elis.sk/index.php?page=shop.product_details&flypage=flypage.tpl&product_id=6164&category_id=150&option=com_virtuemart.
- Nelson MI, Vincent AL. Reverse zoonosis of influenza to swine: new perspectives on the human–animal interface. Trends Microbiol [Internet]. 2015;23:142–153. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0966842X14002467.
- Mena I, Nelson MI, Quezada-Monroy F, et al. Origins of the 2009 H1N1 influenza pandemic in swine in Mexico. Elife [Internet]. 2016;5:e16777. Available from: doi:10.7554/eLife.16777.
- Li Y, Robertson I. The epidemiology of swine influenza. Anim Dis [Internet]. 2021;1:21–25. Available from: doi:10.1186/s44149-021-00024-6.
- Nasamran C, Janetanakit T, Chiyawong S, et al. Persistence of pdm2009-H1N1 internal genes of swine influenza in pigs, Thailand. Sci Rep [Internet]. 2020;10:1–12. Available from: doi:10.1038/s41598-020-76771-2.
- Lam TT, Pybus OG. Genomic surveillance of avian-origin influenza A viruses causing human disease. Genome Med [Internet]. 2018;10:50. Available from: doi:10.1186/s13073-018-0560-3.
- McHardy AC, Adams B. The role of genomics in tracking the evolution of influenza A virus. PLoS Pathog [Internet]. 2009;5:e1000566. Available from: doi:10.1371/journal.ppat.1000566.
- Ospina-Jimenez AF, Gomez AP, Rincon-Monroy MA, et al. Sequence-based antigenic analyses of H1 swine influenza A viruses from Colombia (2008–2021) reveals temporal and geographical antigenic variations. Viruses [Internet]. 2023;15:2030. Available from: doi:10.3390/v15102030.
- Osorio-Zambrano WF, Ospina-Jimenez AF, Alvarez-Munoz S, et al. Zooming in on the molecular characteristics of swine influenza virus circulating in Colombia before and after the H1N1pdm09 virus. Frontiers in Veterinary Science [Internet]. 2022;9:983304. Available from: doi:10.3389/fvets.2022.983304.
- Ciuoderis-Aponte K, Diaz A, Muskus C, et al. Farm management practices, biosecurity and influenza a virus detection in swine farms: a comprehensive study in Colombia. Porcine Health Management [Internet]. 2022;8:42. Available from: doi:10.1186/s40813-022-00287-6.
- Ciuoderis KA, Perez LS, Cardona A, et al. Use of oral fluids for efficient monitoring of influenza viruses in swine herds in Colombia. Revista Colombiana de Ciencias Pecuarias [Internet]. 2022;35:141–152. Available from: https://revistas.udea.edu.co/index.php/rccp/article/view/348325.
- Karlsson EA, Ciuoderis K, Freiden PJ, et al. Prevalence and characterization of influenza viruses in diverse species in Los Llanos, Colombia. Emerg Microbes Infect [Internet]. 2013;2:1–10. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=PMC3636595.
- Ramirez-Nieto GC, Rojas CAD, Alfonso VJV, et al. First isolation and identification of H1N1 swine influenza viruses in Colombian pig farms. Health [Internet]. 2012;04:983–990. Available from: http://www.scirp.org/journal/doi.aspx?DOI=10.4236health.2012.430150.
- Khare S, Gurry C, Freitas L, et al. GISAID’s role in pandemic response. China CDC Weekly [Internet]. 2021;3:1049–1051. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=PMC8668406.
- NCBI. National Center for Biotechnology Information (NCBI) [Internet]. National Center for Biotechnology Information (NCBI). Bethesda (MD): National Library of Medicine (US), National Center for Biotechnology Information. 1988 [cited 2024]. Available from: https://www.ncbi.nlm.nih.gov/.
- Boni MF, de Jong MD, van Doorn HR, et al. Guidelines for identifying homologous recombination events in influenza A virus. PLoS One [Internet]. 2010;5:e10434. Available from: doi:10.1371/journal.pone.0010434.
- Abente EJ, Santos J, Lewis NS, et al. The molecular determinants of antibody recognition and antigenic drift in the H3 hemagglutinin of swine influenza A virus. J Virol [Internet]. 2016;90:8266–8280. Available from: doi:10.1128/jvi.01002-16.
- Walia RR, Anderson TK, Vincent AL. Regional patterns of genetic diversity in swine influenza A viruses in the United States from 2010 to 2016. Influenza Other Respi Viruses [Internet]. 2019;13:262–273. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6468071/.
- Tochetto C, Junqueira DM, Anderson TK, et al. Introductions of human-origin seasonal H3N2, H1N2 and pre-2009 H1N1 influenza viruses to swine in Brazil. Viruses [Internet]. 2023;15:576. Available from: doi:10.3390/v15020576.
- Hanssen H, Hincapié O, López JH. Influenza en porcinos de Antioquia, Colombia. Boletín de la oficina sanitaria panamericana [Internet. 1977;82:35–43. Available from: http://iris.paho.org/xmlui/bitstream/handle/123456789/17291/v82n1p35.pdf?sequence=1.
- Everett HE, Nash B, Londt BZ, et al. Interspecies transmission of reassortant swine influenza a virus containing genes from swine influenza A(H1N1)pdm09 and A(H1N2) viruses. Emerg Infect Dis [Internet]. 2020;26:273–281. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6986826/.
- Pereda A, Rimondi A, Cappuccio J, et al. Evidence of reassortment of pandemic H1N1 influenza virus in swine in Argentina: are we facing the expansion of potential epicenters of influenza emergence? Influenza Other Respi Viruses [Internet]. 2011;5:409–412. Available from: doi:10.1111/j.1750-2659.2011.00246.x.
- Kong W-L, Huang Y-M, Cao N, et al. Isolation and phylogenetic analysis of H1N1 swine influenza virus from sick pigs in southern China. Indian J Virol [Internet]. 2011;22:66–71. Available from: doi:10.1007/s13337-011-0035-2.
- Shortridge KF, Stuart-Harris CH. An influenza epicentre? Lancet [Internet]. 1982;320:812–813. Available from: doi:10.1016/s0140-6736(82)92693-9.
- Panamerican Health Organization. Influenza en porcinos de Antioquia, Colombia [Internet]. Institutional repository for resources sharing. 1977 [cited 2024 Jan 5]. Available from: https://iris.paho.org/bitstream/handle/10665.2/17291/v82n1p35.pdf?sequence=1&isAllowed=y.
- Schweiger B, Zadow I, Heckler R. Antigenic drift and variability of influenza viruses. Med Microbiol Immunol [Internet]. 2002;191:133–138. Available from: https://pubmed.ncbi.nlm.nih.gov/12458347/.
- Chastagner A, Hervé S, Quéguiner S, et al. Genetic and antigenic evolution of European swine influenza A viruses of HA-1C (Avian-Like) and HA-1B (Human-Like) Lineages in France from 2000 to 2018. Viruses [Internet]. 2020;12:1304. Available from: https://www.mdpi.com/1999-4915/12/11/1304.
- He C-Q, Han G-Z, Wang D, et al. Homologous recombination evidence in human and swine influenza A viruses. Virology [Internet]. 2008;380:12–20. Available from: doi:10.1016/j.virol.2008.07.014.
- Bean WJ Jr, Cox NJ, Kendal AP. Recombination of human influenza A viruses in nature. Nature [Internet]. 1980;284:638–640. Available from: doi:10.1038/284638a0.
- Boni MF, Zhou Y, Taubenberger JK, et al. Homologous recombination is very rare or absent in human influenza A virus. J Virol [Internet]. 2008;82:4807–4811. Available from: doi:10.1128/jvi.02683-07.
- Cheung JTL, Lau EHY, Jin Z, et al. Influenza A virus transmission in swine farms and during transport in the swine supply chain. Transbound Emerg Dis [Internet]. 2022;69:e3101–e3110. Available from: doi:10.1111/tbed.14667.
- PorkColombia. Estadisticas Sectoriales [Internet]. Asociacion Colombiana de Porcicultores PorkColombia. 2024 [cited 2024]. Available from: https://porkcolombia.co/estadisticas-sectoriales/.
- Mastin A, Alarcon P, Pfeiffer D, et al. Prevalence and risk factors for swine influenza virus infection in the English pig population. PLoS Curr [Internet]. 2011;3:RRN1209. Available from: doi:10.1371/currents.RRN1209.
- Markin A, Ciacci Zanella G, Arendsee ZW, et al. Reverse-zoonoses of 2009 H1N1 pandemic influenza A viruses and evolution in United States swine results in viruses with zoonotic potential. PLoS Pathog [Internet]. 2023;19:e1011476. Available from: doi:10.1371/journal.ppat.1011476.
- Chauhan RP, Gordon ML. A systematic review of influenza A virus prevalence and transmission dynamics in backyard swine populations globally. Porcine Health Manag [Internet]. 2022;8:10. Available from: doi:10.1186/s40813-022-00251-4.
- Nelson MI, Stratton J, Killian ML, et al. Continual reintroduction of human pandemic H1N1 influenza A viruses into swine in the United States, 2009 to 2014. Sandri-Goldin RM, editor. J Virol [Internet]. 2015;89:6218–6226. Available from: https://jvi.asm.org/content/89/12/6218.
- Cador C, Andraud M, Willem L, et al. Control of endemic swine flu persistence in farrow-to-finish pig farms: a stochastic metapopulation modeling assessment. Vet Res [Internet]. 2017;48:58. Available from: http://veterinaryresearch.biomedcentral.com/articles/10.1186s13567-017-0462-1.
- Sreta D, Tantawet S, Na Ayudhya SN, et al. Pandemic (H1N1) 2009 virus on commercial swine farm, Thailand. Emerg Infect Dis [Internet]. 2010;16:1587–1590. Available from: https://pubmed.ncbi.nlm.nih.gov/20875285/.
- Henritzi D, Petric PP, Lewis NS, et al. Surveillance of European domestic pig populations identifies an emerging reservoir of potentially zoonotic swine influenza A viruses. Cell Host Microbe [Internet]. 2020;28:614–627.e6. Available from: doi:10.1016/j.chom.2020.07.006.
- Rajão DS, Walia RR, Campbell B, et al. Reassortment between swine H3N2 and 2009 pandemic H1N1 in the United States resulted in influenza A viruses with diverse genetic constellations with variable virulence in pigs. Schultz-Cherry S, editor. J Virol [Internet]. 2017;91. Available from: doi:10.1128/JVI.01763-16.
- Peng X, Wu H, Xu L, et al. Molecular characterization of a novel reassortant H1N2 influenza virus containing genes from the 2009 pandemic human H1N1 virus in swine from eastern China. Virus Genes [Internet]. 2016;52:405–410. Available from: https://pubmed.ncbi.nlm.nih.gov/26980674/.
- Rajao DS, Vincent AL, Perez DR. Adaptation of human influenza viruses to swine [Internet]. Frontiers in Veterinary Science. Frontiers Media S.A.; 2019. Available from: https://pubmed.ncbi.nlm.nih.gov/30723723/.
- Chastagner A, Bonin E, Fablet C, et al. Virus persistence in pig herds led to successive reassortment events between swine and human influenza A viruses, resulting in the emergence of a novel triple-reassortant swine influenza virus. Vet Res [Internet]. 2019;50:77. Available from: https://pubmed.ncbi.nlm.nih.gov/31590684/.
- Chou Y-Y, Albrecht RA, Pica N, et al. The M segment of the 2009 new pandemic H1N1 influenza virus is critical for its high transmission efficiency in the guinea pig model. J Virol [Internet]. 2011;85:11235–11241. Available from: https://pubmed.ncbi.nlm.nih.gov/21880744/.
- Tapia R, Torremorell M, Culhane M, et al. Antigenic characterization of novel H1 influenza A viruses in swine. Sci Rep [Internet]. 2020;10:4510. Available from: http://www.nature.com/articles/s41598-020-61315-5.
- Schaefer R, Rech RR, Gava D, et al. A human-like H1N2 influenza virus detected during an outbreak of acute respiratory disease in swine in Brazil. Arch Virol [Internet]. 2015;160:29–38. Available from: https://pubmed.ncbi.nlm.nih.gov/25209152/.
- Anderson CS, McCall PR, Stern HA, et al. Antigenic cartography of H1N1 influenza viruses using sequence-based antigenic distance calculation. BMC Bioinformatics [Internet]. 2018;19:51. Available from: doi:10.1186/s12859-018-2042-4.
- López-Valiñas Á, Valle M, Wang M, et al. Vaccination against swine influenza in pigs causes different drift evolutionary patterns upon swine influenza virus experimental infection and reduces the likelihood of genomic reassortments. Front Cell Infect Microbiol [Internet]. 2023;13:1111143. Available from: doi:10.3389/fcimb.2023.1111143.
- Pan American Health Organization. Epidemiological update: outbreaks of avian influenza caused by influenza A(H5N1) in the region of the Americas [Internet]. Pan American Health Organization. 2023. Available from: https://www.paho.org/en/documents/epidemiological-update-outbreaks-avian-influenza-caused-influenza-ah5n1-region-americas.
- Powdrill TF, Nipp TL, Rinderknecht JL. One health approach to influenza: assessment of critical issues and options 1. Emerg Infect Dis [Internet]. 2010;16:e1–e1. Available from: doi:10.3201/eid1608.100673.
- Saha S, Davis WW. The need for a one health approach for influenza surveillance [Internet]. Lancet Glob. Health. Elsevier BV. 2022;;10::e1078–e1079. Available from: doi:10.1016/S2214-109X(22)00240-6.
- Zheng Z, Lu Y, Short KR, et al. One health insights to prevent the next HxNy viral outbreak: learning from the epidemiology of H7N9. BMC Infect Dis [Internet]. 2019;19:138. Available from: doi:10.1186/s12879-019-3752-6.
- Sommanustweechai A, Iamsirithaworn S, Patcharanarumol W, et al. Adoption of one health in Thailand’s national strategic plan for emerging infectious diseases. J Public Health Policy [Internet]. 2017;38:121–136. Available from: doi:10.1057/s41271-016-0053-9.
- Franklin SI. Can one health fight H5N1 avian influenza? Lancet Planet Health [Internet]. 2023;7:e442–e443. Available from: doi:10.1016/S2542-5196(23)00086-4.
- Ghai RR, Wallace RM, Kile JC, et al. A generalizable one health framework for the control of zoonotic diseases. Sci Rep [Internet]. 2022;12:8588. Available from: doi:10.1038/s41598-022-12619-1.
- Innes GK, Lambrou AS, Thumrin P, et al. Enhancing global health security in Thailand: strengths and challenges of initiating a one health approach to avian influenza surveillance. One Health [Internet]. 2022;14:100397. Available from: doi:10.1016/j.onehlt.2022.100397.
- Roppa L, Duarte ME, Kim SW. — Invited review — pig production in Latin America. Anim Biosci [Internet]. 2024;37:786–793. Available from: doi:10.5713/ab.23.0453.
- Hao R, Liu Y, Shen W, et al. Surveillance of emerging infectious diseases for biosecurity. Sci China Life Sci [Internet]. 2022;65:1504–1516. Available from: doi:10.1007/s11427-021-2071-x.