
ABSTRACT
A positive-sense (+) single-stranded RNA (ssRNA) virus (e.g. enterovirus A71, EV-A71) depends on viral polypeptide translation for initiation of virus replication after entry. We reported that EV-A71 hijacks Hsp27 to induce hnRNP A1 cytosol redistribution to initiate viral protein translation, but the underlying mechanism is still elusive. Here, we show that phosphorylation-deficient Hsp27-3A (Hsp27S15/78/82A) and Hsp27S78A fail to translocate into the nucleus and induce hnRNP A1 cytosol redistribution, while Hsp27S15A and Hsp27S82A display similar effects to the wild type Hsp27. Furthermore, we demonstrate that the viral 2A protease (2Apro) activity is a key factor in regulating Hsp27/hnRNP A1 relocalization. Hsp27S78A dramatically decreases the IRES activity and viral replication, which are partially reduced by Hsp27S82A. However, Hsp27S15A displays the same activity as the wild-type Hsp27. Peptide S78 potently suppresses EV-A71 protein translation and reproduction through blockage of EV-A71-induced Hsp27 phosphorylation and Hsp27/hnRNP A1 relocalization. A point mutation (S78A) on S78 impairs its inhibitory functions on Hsp27/hnRNP A1 relocalization and viral replication. Taken together, we demonstrate the importance of Ser78 phosphorylation of Hsp27 regulated by virus infection in nuclear translocation, hnRNP A1 cytosol relocation, and viral replication, suggesting a new path (such as peptide S78) for target-based antiviral strategy.
GRAPHICAL ABSTRACT
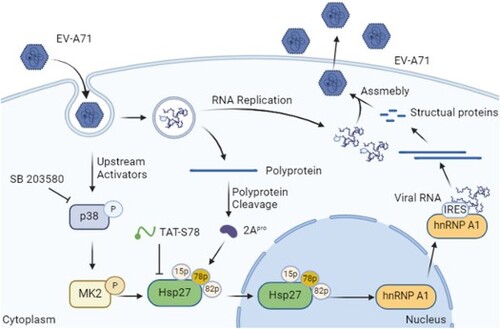
Introduction
Enterovirus A71 (EV-A71), a positive-sense, single-stranded RNA virus [(+) ssRNA virus] in the Picornaviridae family, is regarded as the main aetiological pathogen responsible for human hand-foot-and-mouth disease (HFMD). HFMD is usually a self-limited disease accompanied by some mild symptoms, including fever, exanthema, and oral ulcers [Citation1,Citation2]. It is also one of the most common neurotropic viruses to cause severe central nervous system (CNS) complications, including aseptic meningitis, acute flaccid paralysis, brainstem encephalitis, heart failure, and even death [Citation3,Citation4]. EV-A71 infects millions of children and causes hundreds to thousands of deaths in China annually [Citation5]. In response to the outbreak, China has approved EV-A71 C4 genotype-based vaccines exclusively for children, making it the world's only licensed vaccine for controlling EV-A71 [Citation6,Citation7]. Supportive therapy remains the primary treatment approach for EV-A71 infection, because there is no approved therapeutic drug to combat EV-A71 infections [Citation8].
Positive-sense ssRNA viruses (e.g. picornaviruses, flaviviruses, coronaviruses) rely on the cytosol heterogenous nuclear ribonucleoproteins (hnPNPs) to initiate translation polypeptide for viral replication [Citation9]. After entry and uncoating, EV-A71 initiates its life cycle through an internal ribosome entry site (IRES) directed viral protein translation [Citation10]. The IRES is located in the 5’-UTR of EV-A71, which cap-independently recruits 40S ribosomal components with the help of several host RNA binding proteins known as IRES trans-acting factors (ITAFs) [Citation11]. These ITAFs bind to viral RNA across multiple domains and act to stabilize the structure of IRES when recruiting canonical translation factors and ribosomal subunits [Citation12]. A number of ITAFs have been identified to be involved in EV-A71 and other viruses’ replication, such as hnRNP A1, FBP1, hnRNPK, AUF1, etc. [Citation13,Citation14]. hnRNP A1, a regulator of alternative splicing in the nucleus, is reported to directly bind with IRES as a noncanonical ITAF [Citation9,Citation15], changing the conformation for initiation of viral translation. As a (+) ssRNA virus, EV-A71 completes its life cycle in the cytosol, and the relocalization of hnRNP A1 is of extreme importance for viral protein translation and genome RNA replication [Citation16]. It is reported that viral protease 2Apro or 3Cpro cleaves the component proteins of the nuclear pore complex that may block the trafficking of hnRNP A1 between the nucleus and cytosol in the fast initiation of viral protein translation in this process [Citation17]. We reported that Hsp27 promotes EV-A71 propagation through enhancing viral IRES activity via 2A protease (2Apro)-mediated EIF4G cleavage and hnRNP A1 relocalization from the nucleus to the cytosol [Citation18]. However, the underlying mechanism is elusive.
Hsp27 works as a molecular chaperone in response to various stresses including pathogen infections [Citation1]. Hsp27 plays crucial roles in various types of cancers, inflammatory diseases, neurological diseases, the immune response, and virus infections [Citation19], such as hepatitis B Virus [Citation20], Porcine circovirus type 2 (PCV2) [Citation21], and Dengue virus (DENV) infections [Citation22]. Hsp27 can be modulated by phosphorylation at serine 15, 78, and 82 in response to a variety of signals, including heat shock, oxidative stress, or growth factors [Citation23]. The mitogen-activated protein kinases associated protein kinases (MAPKAP kinases 2, 3), downstream of MAP p38 protein kinase, are responsible for the phosphorylation of Hsp27 [Citation22,Citation24]. Various stimuli, including DNA damage, inflammatory cytokines, and viral infections, can trigger the activation of the p38 MAPK pathway [Citation25,Citation26]. Multiple studies have shown that p38 MAPK signalling was activated by different types of viruses, including EV-A71 [Citation27], SARS-COV-2 [Citation28], ZIKV [Citation29], and Junin Virus (JUNV) [Citation30]. The p38 MAPK inhibitor, SB203580, reduced viral replication and inhibited the secretion of inflammatory factors (such as IL-6, IL-10, and TNF-α), the main cause of viral pathogenesis and death [Citation31]. In this study, we investigated the effects of Hsp27 phosphorylation on hnRNP A1 redistribution triggered by EV-A71 infection or ectopic 2Apro expression and virus replication. We demonstrated the potent antiviral effect of a peptide S78, which targets the key phosphorylation site of Hsp27.
Results
Phosphorylation is required for Hsp27 nuclear localization upon EV-A71 infection
We first investigated the effects of EV-A71 infection on Hsp27 phosphorylation. We showed that EV-A71 infection increased the phosphorylation level of Hsp27 on serine 15 (Ser15), Ser78, and Ser82 (Figure S1). Then we further examined the phosphorylation status of Hsp27 upon virus infection under the treatment with a p38 inhibitor, since the phosphorylation of Hsp27 could be a result of p38 signalling activation [Citation32]. We treated the cells with SB203580 at different concentrations (0, 0.25, 0.5, 1, 2, 4, and 8 µM) and examined the phosphorylation level of Hsp27 upon EV-A71 infection by western blot assay. We observed that SB203580 inhibited the phosphorylation of Hsp27 at different degrees. The phosphorylation level slightly decreased at Ser15 and Ser82. Surprisingly, the phosphorylation at Ser78 was highly sensitive to SB203580 and markedly suppressed in a dose-dependent manner (A). After quantification, with 8 µM of SB203580 treatment, the phosphorylation level at Ser78 reduced by over 85% (Figure S2A).
Figure 1. Requirement of Hsp27 phosphorylation for its nuclear localization upon EV-A71 infection. (A) RD cells were treated with SB203580 at indicated concentration for 2 h, then infected with EV-A71 at the MOI of 10 for 6 h. Cell lysates were collected for western blot assay. Hsp27 phosphorylation on Ser 15, 78, 82 was detected by specific antibodies. GAPDH was detected as internal control. (B) RD cells on the coverslips were first treated with indicated dosage of SB203580 for 2 h, then non-infected or infected with EV-A71 at a MOI of 40 for 6 h. The cells were fixed and stained with anti-Hsp27 (Red), followed by Alexa Flour 594-conjugated anti-rabbit antibody. The nuclei were visualized by staining with DAPI (Blue). The images were captured by Nikon A1HD25 Confocal Microscope. (C) RD cells were treated with SB203580 at indicated concentrations for 2 h, then infected with EV-A71 at the MOI of 10 for 6 h. Cell lysates were collected for nuclear/cytosol fraction assay. Lamin B1 was detected as internal nuclear control. GAPDH was detected as internal cytosol control.
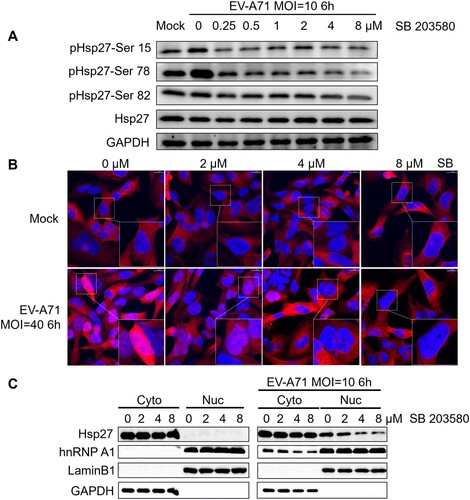
Hsp27 is predominantly located in the cytoplasm [Citation33]. However, stress (e.g. heat shock, virus infection) can induce partial redistribution of Hsp27 from the cytoplasm to the nucleus, concomitantly with its phosphorylation [Citation34,Citation35]. HSV-1 infection induced the redistribution of Hsp27 in VERO cells [Citation36]. We postulated that the cellular redistribution of Hsp27 may be associated with its phosphorylation upon EV-A71 infection. We treated RD cells with SB203580 at 0, 2, 4, and 8 µM for 2 h, and then infected the cells with EV-A71 for 6 h. After immunostaining, we observed that Hsp27 was not translocated to the nucleus without EV-A71 infection, regardless of whether the cells were treated with SB253080 (B, upper panels). Upon EV-A71 infection, a large amount of Hsp27 was translocated from cytosol to nucleus without SB253080 treatment (B, the left lower panel), while a markedly reduced nuclear Hsp27 level was observed when the cells were treated with 2 µM of SB253080. When the inhibitor concentration was increased to 4 µM, the nuclear relocation of Hsp27 was further blocked to an invisible level (B, lower panels). The colocalization of Hsp27 and DAPI, which stands for the level of redistribution of Hsp27, was also quantified. The colocalization coefficient M1 was computed as the fraction of Hsp27 colocalized with DAPI in the total Hsp27 area using the JACoP-plugin in FIJI. Without SB203580 treatment, the fraction of Hsp27 overlapping DAPI was 45%, while it was reduced in a dose-dependent manner. At 8 µM SB203580 treatment, the fraction of Hsp27 overlapping DAPI was even reduced to the basal level (Figure S2B). Moreover, we treated RD cells with SB203580 at 0, 2, 4, and 8 µM for 2 h, and then infected with EV-A71 for 6 h. After separately extracting the nuclear and cytoplasm protein, we found that Hsp27 locates in cytoplasm in RD cells. Upon EV-A71 infection, Hsp27 dramatically translocates into nucleus. As expected, SB203580 treatment can suppress the nuclear translocation of Hsp27 in a dose-dependent manner (C).
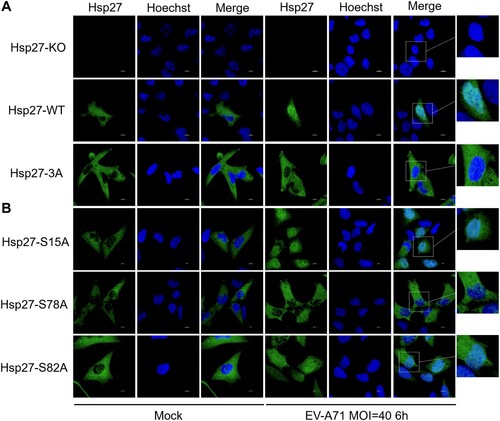
To further confirm the role of Hsp27 phosphorylation in its nuclear translocation, we constructed lentivirus vectors to restore the wild-type (WT) Hsp27 and phosphorylation-deficient mutant (Hsp27S15/78/82A, Hsp27-3A) in Hsp27 knockout RD cells (Hsp27-KO RD cells) for three days. After EV-A71 infection at the MOI (multiplicity of infection) 40 for 6 h, we examined the location of Hsp27 using an immunofluorescence assay. The restored Hsp27 relocated from the cytoplasm into the nucleus 6 h post-infection (p.i.). However, little nuclear Hsp27-3A was observed, indicating the importance of Hsp27 phosphorylation on its nuclear translocation (A).
Figure 2. A critical role of Ser78 phosphorylation in the Hsp27 nuclear localization upon EV-A71 infection. (A–B) Hsp27-KO RD cells on the coverslips were infected with Lenti-Hsp27-WT, Lenti-Hsp27-3A, Lenti-Hsp27-S15A, Lenti-Hsp27-S78A, or Lenti-Hsp27-S82A for 3 days, then infected with EV-A71 at the MOI of 40 for 6 h. The cells were fixed and stained with anti-Hsp27 (Green), followed by Alexa Fluor 488-conjugated anti-rabbit antibody. The nuclei were stained with Hoechst (Blue). The images were captured by Nikon A1HD25 Confocal Microscope.
Ser78 phosphorylation plays a crucial role in Hsp27 nuclear localization upon EV-A71 infection
To figure out the key phosphorylation site contributing to Hsp27 nuclear localization, phosphorylation-deficient Hsp27 mutants (Hsp27S15A, Hsp27S78A, and Hsp27S82A) were restored in Hsp27-KO RD cells with lentivirus vectors for three days. The localization of the Hsp27 mutants was captured after EV-A71 infection for 6 h. We showed that both Hsp27S15A and Hsp27S82A relocated from the cytoplasm into the nucleus, while little Hsp27S78A displayed in the nucleus (B). These results indicated that the Ser78 phosphorylation is critical for Hsp27 nuclear localization upon EV-A71 infection.
Ser78 phosphorylation of Hsp27 is critical for induction of hnRNP A1 cytosol redistribution upon EV-A71 infection
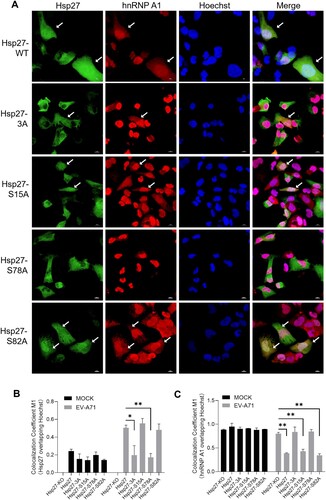
As shown in C, SB 203580 inhibited the cytosol translocation of hnRNP A1 in a dose-dependent manner upon EV-A71 infection. To further dissect the effects of Hsp27 phosphorylation on hnRNP A1 cytosol redistribution, the wild type Hsp27 or a phosphorylation-deficient Hsp27 mutant (Hsp27-3A, Hsp27S15A, Hsp27S78A, and Hsp27S82A) were restored in Hsp27-KO RD cells for 3 days, which were then infected with EV-A71 for 6 h. As shown in A, the cytosol redistribution of hnRNP A1 was completely blocked in the cells with the restoration of Hsp27S78A expression but successfully rescued in the cytosol after the restoration of Hsp27S15A and Hsp27S82A expression. Results from quantification of colocalization showed that both Hsp27 and the phosphorylation-deficient Hsp27 mutants were mostly localized in the cytosol but not in the nucleus without EV-A71 infection; meanwhile, hnRNP A1 localized in the nucleus without cytosol distribution. Upon EV-A71 infection, a certain portion of Hsp27, Hsp27S15A, and Hsp27S82A translocated into the nucleus, and accordingly, hnRNP A1 was obviously redistributed in the cytosol. However, in Hsp27-3A or Hsp27S78A-restored cells, the level of colocalization between Hsp27 and Hoechst failed to be increased by EV-A71 infection (B), and most hnRNP A1 was still colocalized with Hoechst (C). Taken together, the Ser78 phosphorylation of Hsp27 displayed a key role in both Hsp27 nuclear translocation and hnRNP A1 cytosol redistribution upon EV-A71 infection.
Figure 3. The requirement of Ser78 phosphorylation of Hsp27 for induction of hnRNP A1 cytosol redistribution upon EV-A71 infection. (A) Hsp27-KO RD cells on the coverslips were infected with Lenti-Hsp27-WT, Lenti-Hsp27-3A, Lenti-Hsp27-S15A, Lenti-Hsp27-S78A, or Lenti-Hsp27-S82A at the MOI of 20 for 3 days, then infected with EV-A71 at the MOI of 40 for 6 h. The cells were fixed and stained with anti-Hsp27 (Green) and anti-hnRNPA1 (Red), followed by Alexa Fluor 488-conjugated anti-rabbit antibody and Alexa Fluor 594-conjugated anti-mouse antibody. The nuclei were stained with Hoechst (Blue). The images were captured by Nikon A1HD25 Confocal Microscope. Cells with translocation were marked by white arrows. (B) Colocalization analysis of C, and this figure was conducted using the JACoP-plugin of the extended ImageJ version Fiji. The M1 colocalization coefficient (the fraction of Hsp27 in Hoechst) was computed. (C) The fraction of hnRNP A1 in Hoechst was quantified in Hsp27 restoring cells with or without EV-A71 infection. Statistical analyzes were carried out using Student’s t-test. *p < 0.05, **p < 0.01. Data are expressed as mean ± SD.
Furthermore, we examined if the phosphorylated Hsp27 itself could direct its redistribution into the nucleus and induce hnRNP A1 cytosol redistribution without EV-A71 infection. It was reported that the replacement of serine with aspartate functions as a phosphorylation-active mimic [Citation37]. As shown in Figure S3A, Hsp27 or phosphorylation-active mimics (Hsp27-3D, Hsp27S15D, Hsp27S78D, and Hsp27S82D) did not translocate from the cytosol to the nucleus; meanwhile, they were not able to induce hnRNP A1 redistribution from the nucleus to cytosol without EV-A71 infection (Figure S3A). On the other hand, the phosphorylation-active Hsp27 mimics are mainly located in the nucleus upon EV-A71 infection; and obviously, hnRNP A1 redistributed in the cytosol upon EV-A71 infection, as the quantification analysis showed (Figure S3B-C). Thus, phosphorylation of Hsp27 cannot directly drive the translocation of hnRNP A1 from the nucleus to cytosol without EV-A71 infection.
2Apro Protease activity is essential for Hsp27-Ser78 phosphorylation-mediated hnRNP A1 cytosol redistribution
Our previous study revealed that Hsp27 promotes EV-A71 infections through 2Apro-enhanced viral IRES activity [Citation18]. To further dissect the relationships among 2Apro, Hsp27 phosphorylation, and hnRNP A1 cytosol redistribution, we restored Hsp27 and its phosphorylation-deficient mutants and then transfected a plasmid to express 2Apro in Hsp27-KO cells. We observed that hnRNP A1 was fully localized in the nucleus in the control cells. 2Apro successfully restored the cytosol redistribution of hnRNP A1 in the cells with Hsp27S15A and Hsp27S82A expression but failed to do so in the cells with Hsp27S78A and Hsp27-3A expression (A, S4A & C). Accordingly, 2Apro obviously directed the cytosol redistribution of hnRNP A1 by phosphorylation-active Hsp27 mimics (Hsp27S15D, Hsp27S78D, Hsp27S82D, and Hsp27-3D) in the Hsp27-KO RD cells (A, S4B & D).
Figure 4. 2Apro protease activity is essential for Hsp27-Ser78 phosphorylation-mediated hnRNP A1 cytosol redistribution. (A–B) Hsp27-KO RD cells on the coverslips were infected with Lenti-Hsp27-WT, Lenti-Hsp27-3A, Lenti-Hsp27-S15A, Lenti-Hsp27-S78A, Lenti-Hsp27-S82A, Lenti-Hsp27-3D, Lenti-Hsp27-S15D, Lenti-Hsp27-S78D, or Lenti-Hsp27-S82D at the MOI of 20 for 3 days, then transfected with 500 ng 2Apro or 2AC110A for 24 h. The cells were fixed and stained with anti-Hsp27 (Green) and anti-hnRNPA1 (Red), followed by Alexa Fluor 488-conjugated anti-rabbit antibody and Alexa Fluor 594-conjugated anti-mouse antibody. The nuclei were stained with Hoechst (Blue). The images were captured by Nikon A1HD25 Confocal Microscope. Cells with translocation were marked by white arrows.
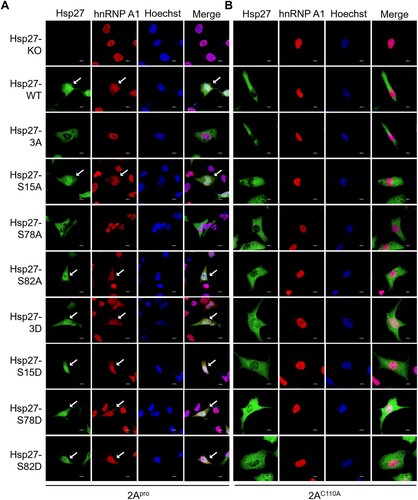
To further examine the effect of protease activity on Hsp27 nuclear translocation and hnRNP A1 cytosol redistribution, the protease-deficient 2AC110A mutant was co-expressed with either wild-type Hsp27, phosphorylation-deficient Hsp27 mutants or phosphorylation-active Hsp27 mimics in the Hsp27-KO RD cells. Interestingly, 2AC110A not only failed to induce Hsp27 translocation from the cytosol to the nucleus, but also was not able to direct the redistribution of hnRNP A1 from the nucleus to the cytoplasm (B, S4), demonstrating an essential role of protease activity in 2Apro for regulation of hnRNP A1 translocation.
Ser78 phosphorylation of Hsp27 is crucial for enhancing viral IRES activity
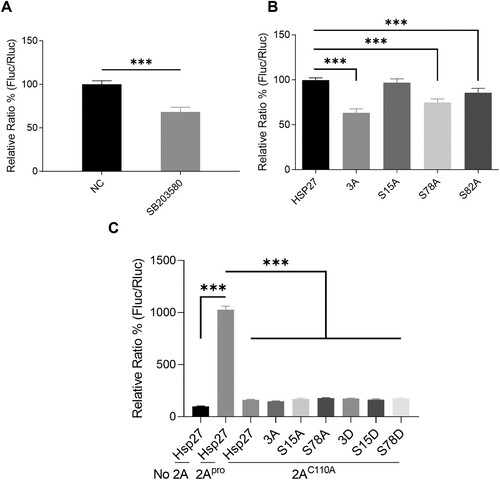
We further examined the effects of Ser78 phosphorylation on viral IRES activity. We first treated RD cells with SB203580 and co-transfected 2Apro-expressing and IRES-driven dicistronic reporter plasmids for 24 h. The renilla luciferase (RLuc) and firefly luciferase (FLuc) activities were determined with a dual-luciferase assay kit. The normalized ratio of Fluc to Rluc presented the relative IRES activity. After pretreatment of RD cells with 2 μM of SB203580, the IRES activity was reduced by 32% (A). Since HEK293 T cells express little endogenous Hsp27, which is even undetectable by western blot as shown by Dan et al. [Citation18], we examined the effects of phosphorylation-deficient Hsp27 on IRES activity in HEK293 T cells. Phosphorylation-deficient Hsp27 expressing plasmids were co-transfected with 2Apro-expressing and pIRES reporter plasmids into HEK293 T cells for 24 h. Similar to SB203580, Hsp27-3A decreased the IRES activity by 37%. Strikingly, Ser78 phosphorylation-deficient Hsp27 (Hsp27S78A) alone reduced the IRES activity by 25%, while Hsp27S82A only marginally reduced the IRES activity (B). Surprisingly, Hsp27S15A displayed no effects on the IRES activity. Furthermore, we examined the IRES activity of Hsp27-restoring cells with or without expression of 2Apro or the inactive 2Apro (2AC110A). We showed that 2Apro increased IRES activity by 10 folds, but 2AC110A displayed almost no effects on the IRES activity in the cells expressed with the wild type Hsp27. Importantly, 2AC110A had no effects on the IRES activity in the cells expressing phosphorylation-deficient or phosphorylation-mimic Hsp27 mutants (C), again demonstrating that phosphorylated Hsp27 cannot induce IRES-dependent protein translation without the help of 2Apro or EV-A71. Taken together, the Ser78-phosphorylation of Hsp27 is a key event to direct the hnRNP A1 cytosol relocalization and facilitate IRES-dependent translation upon EV-A71 infection.
Figure 5. The inhibitory effects of Ser78 phosphorylation-deficient Hsp27 on the viral IRES activity. (A) RD cells were pretreated with 2 μM SB203580 for 2 h, then co-transfected with a 2Apro-expressing plasmid (200 ng) and pIRES reporter plasmid (200 ng) for 24 h, then the luciferase activity was measured by BioTek Synergy H1 microplate reader. (B) HEK 293 T cells were co-transfected with Hsp27 (WT, 3A, S15A, S78A, or S82A) plasmid (800 ng), pIRES reporter plasmid (200 ng) and 2Apro-expressing plasmid (200 ng) for 24 h, then the luciferase activity was measured. (C) HEK 293 T cells were co-transfected with Hsp27 (WT, 3A, S15A, S78A, 3D S15D, or S78D) plasmid (800 ng), pIRES reporter plasmid (200 ng) and 2Apro-expressing plasmid or 2AC110A-expressing plasmid (200 ng) for 24 h, then the luciferase activity was measured. Statistical analyzes were carried out using Student’s t-test. *p < 0.05, **p < 0.01, ***p < 0.001. Data are expressed as mean ± SD.
Ser78 phosphorylation-deficient Hsp27 inhibits EV-A71 replication and propagation
To explore the effects of Hsp27 phosphorylation on EV-A71 replication, Hsp27 and its phosphorylation-deficient mutants were ectopically expressed in 293 T cells for 48 h, which were then infected with EV-A71 at the MOI of 1 for 9 h. The viral RNA level was measured by RT-qPCR. Compared with the control group in which Hsp27 was over-expressed, the viral RNA level decreased by 80% and 50% in cells ectopically expressed Hsp27-3A and Hsp27S78A mutants, respectively (A). We noticed that the viral RNA level only slightly reduced in the Hsp27S82A-expressing cells, whereas no difference of viral RNA levels was observed between the control and Hsp27S15A-expressing groups (A). Accordingly, the viral RNA levels significantly increased about 150%, 90%, and 100% in the groups with ectopic expression of phosphorylation-active Hsp27 mimics Hsp27-3D, Hsp27S78D, and Hsp27S82D respectively (B). Furthermore, we tested the effects of SB203580 on viral replication in the cells ectopic expressing Hsp27. As shown in C, SB203580 decreased the viral RNA level by 50% in the Hsp27 over-expressing cells, but no obvious effects were obtained in the cells with Hsp27S78A- and Hsp27S78D-expressing cells. Again, these results demonstrated the crucial role of Ser78 phosphorylation in viral RNA replication, consistence with its nuclear localization and induction of hnRNP A1 cytosol redistribution.
Figure 6. The inhibitory effects of Ser78 phosphorylation-deficient Hsp27 on EV-A71 replication and propagation. (A) HEK 293 T cells were transfected with 500 ng Hsp27 (WT, 3A, S15A, S78A, or S82A) plasmids for 48 h, and then infected with EV-A71 at an MOI of 10 for 9 h. Intracellular viral RNA was extracted and quantified by qPCR. (B) HEK 293 T cells were transfected with 500 ng Hsp27 (WT, 3D, S15D, S78D, or S82D) plasmids for 48 h, and then infected with EV-A71 at an MOI of 10 for 9 h. Intracellular viral RNA was extracted and quantified by qPCR. (C) HEK 293 T cells were transfected with 500 ng Hsp27 (WT, S78A, or S78D) plasmids for 48 h, and pretreated with 8 μM SB203580 2 h before infection with EV-A71 at an MOI of 10 for 9 h. D. HEK 293 T cells were transfected with 500 ng Hsp27 (WT, 3A, S15A, S78A, or S82A) plasmids for 48 h, and then infected with EV-A71 at an MOI of 10 for 12 h. Viral titration was calculated by TCID50. Statistical analyzes were carried out using Student’s t-test. *p < 0.05, **p < 0.01, ***p < 0.001. Data are expressed as mean ± SD.
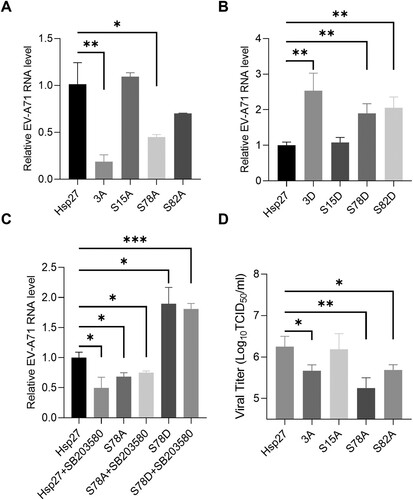
To examine the effects on viral propagation, we over-expressed Hsp27 and phosphorylation-deficient Hsp27 mutants (Hsp27-3A, Hsp27S15A, Hsp27S78A, and Hsp27S82A) in HEK293 T cells, and then infected with EV-A71. Our results showed that the Hsp27-3A and Hsp27S82A reduced the viral titre by 75%, while the viral titre was not reduced by Hsp27S15A. Strikingly, Hsp27S78A reduced the viral titre by over 90% (D).
Peptide S78 suppresses EV-A71 induced Hsp27/hnRNP A1 relocalization and viral IRES activity
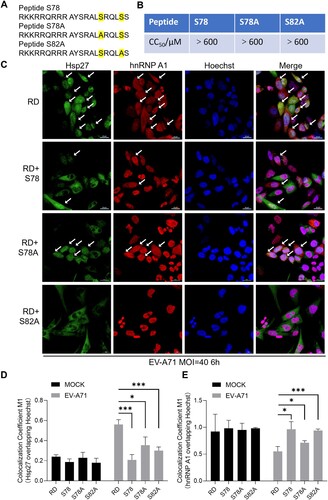
To further demonstrate the antiviral potential by targeting Ser78 phosphorylation, we designed three peptides (residues 71–83 of Hsp27) tagged with TAT sequence for membrane penetration (A). It was supposed that peptides S78 and S82A can interfere Ser78 phosphorylation while peptides S78A (Ser78 replaced by Ala78) would not affect the Ser78 phosphorylation of Hsp27. The CC50 of these peptides was over 600 μM (B), indicating no toxicity of these peptides in the test cells. RD cells were pre-treated with peptide S78, S78A or S82A at the concentrations of 25 μM for 2 h and then infected with or without EV-A71 at an MOI of 40 for 6 h. As shown in Figure S6A, the localization of Hsp27 and hnRNP A1 in mock RD cells was not affected by the peptide treatment. In the EV-A71-infected RD cells, the redistribution of both Hsp27 and hnRNP A1 were dramatically blocked by S78 and S82A treatment but only partially reduced by S78A (C–E). We also examined the effects of 2Apro on the induction of Hsp27 and hnRNP A1 cellular redistribution. In the peptide S78 or S82A-treated RD cells, the 2Apro-induced relocalization of Hsp27 and hnRNP A1 was almost completely blocked, while S78A-treated cells still showed a certain cellular redistribution of both Hsp27 and hnRNP A1 (Figure S5A). The quantification analysis further confirmed our findings (Figure S5B-C). These results indicated that Ser82 displayed less important role in 2Apro-induced Hsp27 and hnRNP A1 relocalization. Consistent with B, 2AC110A did not induce the re-localization of Hsp27 and hnRNP A1 that was also not affected by any peptide treatment (Figure S6B). Two control peptides (NC, and S78-3A, Figure S7A) were used to exclude potential nonspecific effects caused by peptide treatment. These peptides are also not toxic because they showed high CC50 (Figure S7B). NC treatment did not affect EV-A71-induced Hsp27 and hnRNP A1 translocation, but S78-3A partially suppressed their redistribution as shown in the quantitative assay (Figure S7C–E).
Figure 7. Inhibition of Hsp27/hnRNP A1 relocalization by peptide S78. (A) The amino acids sequence of peptide S78, peptide S78A, and peptide S82A. (B) CC50 of the peptides. (C) RD cells on the coverslips were treated with peptides (S78, S78A and S82A) at the concentration of 25 μΜ for 2 h, and then infected with EV-A71 for 6 h at the MOI of 40. The cells were fixed and stained with anti-Hsp27 (Green) and anti-hnRNPA1 (Red), followed by Alexa Fluor 488-conjugated anti-rabbit antibody and Alexa Fluor 594-conjugated anti-mouse antibody. The nuclei were stained with Hoechst (Blue). The images were captured by Nikon A1HD25 Confocal Microscope. Cells with translocation were marked by white arrows. (D) Colocalization analysis of (C) was conducted using the JACoP-plugin of the extended ImageJ version Fiji. The M1 colocalization coefficient (the fraction of Hsp27 in Hoechst) was computed. (E) The fraction of hnRNP A1 in Hoechst was quantified. Statistical analyzes were carried out using Student’s t-test. *p < 0.05, **p < 0.01, ***p < 0.001. Data are expressed as mean ± SD.
The phosphorylated level at Ser15, 78, or 82 of Hsp27 was detected by specific antibodies in the EV-A71 infected cells. S78 treatment dramatically inhibited Ser78 phosphorylation rather than Ser15 or Ser82, while S78A peptide showed less inhibition on Ser78 phosphorylation (A–B). Furthermore, reporter assay showed that 2Apro-induced viral IRES activity was decreased 34% in the S78-treated cells, similar to the level of SB203580 treatment (A). However, S78A treatment only slightly reduced about 10% of the IRES activity (C). As a negative control, NC did not affect viral IRES activity. The reduction in viral IRES activity by S82A treatment was similar to S78, while S78-3A mildly decreased the IRES activity (Figure S8A), indicating the critical role of Ser78 phosphorylation for facilitating viral IRES-dependent protein translation.
Figure 8. Inhibition of EV-A71 replication and propagation by peptide S78. (A) RD cells were pre-treated with peptides S78 or S78A for 2 h and infected with EV-A71 at the MOI of 10 for 6 h. Cell lysates were collected for western blot assay. Hsp27 phosphorylation on Ser 15, 78, 82 were detected by specific antibodies. β-Actin were detected as internal control. (B) Densitometric analysis of (A). (C) HEK 293 T cells were pre-treated with peptides S78 or S78A at the concentration of 25 μΜ for 2 h and then co-transfected with Hsp27-WT plasmid (800 ng), pIRES reporter plasmid (200 ng) and 2Apro-expressing plasmid or 2AC110A-expressing plasmid (200 ng) for 24 h, then the luciferase activity was measured. (D–E) RD cells were treated with peptides S78 or S78A and then infected with EV-A71. EV-A71 VP1 were detected. (F) RD cells were treated with peptides and then infected with EV-A71 at the MOI of 1 for 12 h. The EV-A71-induced CPE was captured. (G–H) Intracellular viral RNA was extracted and quantified by qPCR. (I) Densitometric analysis of (D–E). The analysis used respective untreated control for S78 and S78A. (J) RD cells were treated with peptides S78 or S78A and then infected with EV-A71 at the MOI of 1 for 12 h. Extracellular viral RNA was extracted and quantified by qPCR. (K) Viral titration was calculated by TCID50. The Statistical analyzes were carried out using Student’s t-test. *p < 0.05, **p < 0.01, ***p < 0.001. Data are expressed as mean ± SD.
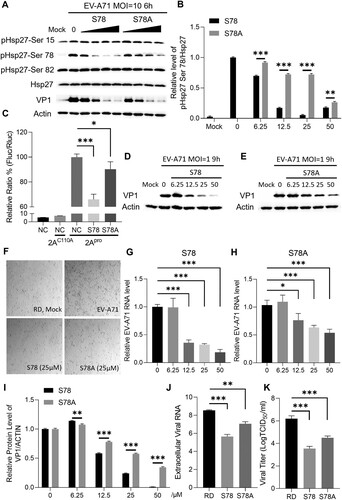
Peptide S78 inhibits EV-A71 replication and propagation
To further explore the potential antiviral effects of peptides S78 on EV-A71 infection, cytopathic effects (CPE) were examined on RD cells infected with EV-A71. S78 treatment dramatically protected the infected cells from EV-A71 infection, similar to the uninfected healthy RD cells, while S78A treatment showed mild protection from EV-A71-induced CPE (F). Moreover, the EV-A71 VP1 protein level was dramatically decreased by S78 in a dose-dependent manner. VP1 was even eliminated to the almost undetectable level at the concentration of 50 μM (D). However, the reduction of VP1 level was much weaker with S78A and S78-3A treatment (E, Figure S8D). Compared to S78, S82A treatment showed a little bit weaker inhibition on VP1 expression (Figure S8C). NC treatment had no effect on VP1 level (Figure S8E). Densitometric assay showed that S78 and S82A reduced VP1 level by over 99% and 90% respectively, while S78A and S78-3A only decreased by about 60% at the concentration of 50 μM (I, S8F). S78 also dramatically reduced the viral RNA in a dose-dependent manner. At the concentration of 50 μM, the viral RNA was decreased by 82% (G), while S78A only reduced the viral RNA by 46% (H), while the NC peptide treatment had no effect on the viral RNA level (Figure S8B). The S78 treatment decreased the extracellular viral RNA level by 800 folds at 25 μM, but S78A only reduced by 30 folds (J). The viral titre decreased 450 folds by S78 treatment but only 50 folds by S78A treatment ( K). S82A treatment had a similar effect on viral titre as compared to S78, while S78-3A only mildly decreased the viral titration (Figure S8G). Taken together, the results further demonstrated that Ser78 phosphorylation plays a crucial role in facilitating EV-A71 infections and targeting Ser78 phosphorylation by S78 and S82A peptides displayed much stronger antiviral effects as compared with S78A peptide. S78 peptide could be a potential candidate for anti-EV-A71 drug development.
Discussion
In response to different types of virus infections, chaperon proteins (e.g. HSPs, hnRNPs) may participate in every step of the viral infections, including entry, uncoating, replication, assembly, and releasing and even innate immunity [Citation1]. SARS-CoV-2 (3CLpro) and EV71 (2Apro) trigger chaperon protein LRPAP1 upregulation and secretion, and then binding to IFNAR1, leading to IFNAR1 degradation for facilitating virus to escape from the host defense [Citation38]. Heat shock proteins (HSPs) are induced by various stress, such as high temperature, starvation, pathogen infection, UV, and chemicals treatment. The viral entry of DENV and JEV is supported by Hsp90 and Hsp70s in microglial cells [Citation39]. Hsp90 inhibitors can even block the EV-A71 entry [Citation40]. Hsp90 can also stabilize the viral protein and modulate the viral polymerase activity, such as NS3 of HCV, to benefit viral replication [Citation41]. EV-A71-induced PIM1 could promote viral replication by activating Hsp90 [Citation1, Citation42]. Upon EV-A71 infection, viral IRES-dependent translation is enhanced by Hsc70, which can also interact with 2Apro to enhance EIF4G cleavage to block host cap-dependent translation [Citation43]. Grp78 suppresses HBV replication through induction of IFNβ expression [Citation44]. Hsp27, one of the most important small HSPs, responds to many virus infections. It has been reported that many viruses can stimulate Hsp27 phosphorylation, including PCV2 [Citation21], HSV [Citation45], EBV [Citation46], and PRRSV [Citation47]. In the uninfected PK-15 cells, there is a diffuse pattern of Hsp27 throughout the cells. After PCV2 infection, Hsp27 accumulates in the nucleus [Citation21]. Hsp27 redistributes from the cytoplasm to the perinuclear region after Sindbis Virus infection [Citation48]. EV-A71 infection upregulates Hsp27 expression to enhance viral IRES activity through 2Apro-induced EIF4G cleavage and hnRNP A1 cytosol translocation [Citation18]. Hsp27 is predominantly located in the cytoplasm in normal cells. The Hsp27 relocalization may be a key event for facilitating viral replication. However, the potential function of Hsp27 phosphorylation and redistribution are not well understood in the viral infections.
In this study, we applied p38 kinase inhibitor SB203580, phosphorylation-deficient mutants, and phosphorylation-active mimics of Hsp27 to demonstrate the functions of phosphorylation in Hsp27 nuclear translocation and hnRNP A1 cytosol redistribution, and the effects on viral IRES activity and viral propagation as well. We observed that EV-A71 infection stimulated the phosphorylation of Hsp27 at Ser15, 78, and 82 (Figure S1) and SB203580 predominantly suppressed Hsp27 phosphorylation at Ser78 in a dose-dependent manner upon EV-A71 infection (A, S2A). We observed that Ser15-phosphorylation-deficient Hsp27S15A mutant did not affect Hsp27/hnRNP A1 redistribution and EV-A71 replication (, 6A), while Ser82-phosphorylation-deficient mutant affected viral IRES activity and replication at a certain degree but had no effects on Hsp27/hnRNP A1 redistribution (, 5B, 6A). We assume that Ser82 phosphorylation may contribute to EV-A71 infection with some unknown mechanisms. To our surprise, Ser78 phosphorylation-deficient Hsp27S78A mutant alone lost the ability to direct Hsp27/hnRNP A1 relocalization upon EV-A71 infection, reduced IRES activity and potently inhibited EV-A71 replication and propagation (–3, 5B, 6). All these results demonstrated a crucial role of Ser78 phosphorylation in facilitating EV-A71 infections.
To evade host antiviral response and disrupt the nuclear transport pathway, enterovirus 2Apro cleaves diverse host proteins, such as eIF4G, P-body, PABP, TRAF3, MAVS, IFNAR1, and nucleoporins (Nups) [Citation49–54]. In this study, 2Apro successfully induced Hsp27 nuclear translocation and hnRNP A1 cytosol redistribution in Hsp27-restoring cells (A, S4). Considering the crucial role of phosphorylation for Hsp27 nuclear translocation, 2Apro would be sufficient to induce Hsp27 phosphorylation at Serine 78. However, 2AC110A cannot elicit Hsp27 nuclear translocation or hnRNP A1 cytosol redistribution in phosphorylation-mimic Hsp27 mutants-restoring cells (B, S4). Therefore, besides the Hsp27 phosphorylation, the protease activity is of importance for Ser78-phosphorylated Hsp27 nuclear translocation and hnRNP A1 cytosol redistribution. The nuclear import of Hsp27 may require the involvement of other host proteins which may be cleaved by 2Apro.
Currently, there is no FDA-approved antiviral drug for the treating enterovirus infected patients. Although some compounds have been shown antiviral effects against EV-A71 infection [Citation55,Citation56], the agents still face numerous challenges such as adverse side effects and off-target toxicity [Citation57,Citation58]. Importantly, peptides have great advantages over small molecular agents because of their high selectivity, low/no side effects, and commercial scalability [Citation59]. We therefore further investigated the antiviral potential by targeting Ser78 phosphorylation through peptide competition. S78 peptide shares the same sequence as Hsp27. It should be a strong competitor of Ser78 phosphorylation of Hsp27. Peptide S82A should also a strong inhibitor of Ser78 phosphorylation of Hsp27 because the Ser78 keeps intact. S78A peptide should not have strong effective activity to inhibit Ser78 phosphorylation of Hsp27 since Ser78 is replaced with Ala78 (A). We showed that the Hsp27/hnRNP A1 redistribution was potently suppressed by S78 peptide, while the blockage was much weaker by S78A treatment in the EV-A71 infected cells (). We also showed that S78 dramatically inhibited Ser78 phosphorylation of Hsp27 in a dose-dependent manner, while S78A displayed much less inhibitory effects (). S78 treatment decreased the 2Apro-induced IRES activity by 34%, which was marginally reduced by S78A (C). More importantly, S78 treatment amusingly reduced the viral protein VP1 and intracellular RNA level by 99% and 82%, which could be rescued by one site mutation on S78 (S78A) (D–I). S78 treatment was able to decrease viral titre by 2.7 logs ( K), demonstrated a great potential for antiviral treatment in clinical applications. Results from our study reveal a specific mechanism of Hsp27-derived peptide S78 against EV-A71 infection, paving a way for developing target-specific antiviral peptide treatment. Recent studies have shown the promising application of supramolecular adhesive materials with antimicrobial activity [Citation60,Citation61]. Combined with a target-specific antiviral drug, the adhesive materials may have a huge potential for target-specific and tissue-specific treatment for EV-A71 infection.
In summary, EV-A71 infection causes Hsp27 phosphorylation at three residues (Ser15, Ser78, and Ser82). SB203580 mainly inhibited Hsp27 phosphorylation at Ser78 and corresponding EV-A71 replication. The phosphorylation at serine 78 of Hsp27 plays a critical role in the process of Hsp27 nuclear translocation, hnRNP A1 cytosol redistribution, viral protein translation, and replication upon EV-A71 infection. However, the blockage of hnRNP A1 relocalization cannot be induced by phospho-mimic Hsp27 mutants without EV-A71 infection. 2Apro but not 2AC110A successfully induces the translocation of Hsp27 and hnRNP A1 in wild-type RD cells but not in the Hsp27-3A or Hsp27S78A restored cells, indicating the essential role of 2Apro in this mechanism. Although Ser82 phosphorylation has certain effects on viral protein translation and replication, it has little effects on the Hsp27 nuclear translocation and hnRNP A1 cytosol redistribution. Interestingly, the Ser15 phosphorylation plays no role in any of this process. Peptide (S78) dramatically suppresses 2Apro-induced the relocalization of Hsp27 and hnRNP A1, viral IRES activity, viral protein translation, and viral reproduction, indicating that peptide S78 is an ideal drug candidate for the treatment of EV-A71 infection.
Materials and methods
Cells and virus
HEK293 T cells and RD cells (ATCC #CCL-136) were cultured in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum (FBS) and supplemented with 100U/mL of penicillin and 100 µg/mL of streptomycin. Hsp27 knockout RD cells (Hsp27-KO RD cells) were previously constructed using CRISPR/Cas 9 and maintained in the lab [Citation18]. The targeting sgRNA sequence was 5’-GCAUAGCCGCCUCUUCGACC-3’. EV-A71 was obtained from Shenzhen Center for Disease Control and Prevention (SHZH98 strain, GenBank accession number AF302996) and propagated as previously described [Citation62]. The virus was aliquoted and stored at −80°C until use.
Plasmids
The mutants of Hsp27 (Hsp27-3A, S15A, S78A, S82A, 3D, S15D, S78D, and S82D) were constructed using a one-step mutagenesis kit (GeneTailor Site-Directed Mutagenesis System, Invitrogen, USA) on pcDNA4/HisMax B-Hsp27. The correction of constructs was confirmed by automated DNA sequences. The EV-A71 IRES reporter plasmid, pcDNA4/HisMax B-2Apro, and pcDNA4/HisMax B-2AC110A were constructed as previously described [Citation43,Citation52]. The protease-inactive mutation of 2Apro (Cys110 to Ala110, 2AC110A) was generated by site-directed mutagenesis with a one-step mutagenesis kit (Invitrogen). To construct EV-A71 IRES reporter plasmid, the Renilla Luciferase gene (RLuc) was inserted into pcDNA4/HisMax B between BamH I and EcoR V sites first, then the amplified IRES-FLuc encoding sequence was inserted downstream of the RLuc using EcoR V and Xbar I. The primers used in amplification are available upon request.
Lentivirus package
Lentiviral vectors were prepared as described previously [Citation63]. Briefly, The GFP sequence of pLVTHM was substituted with the sequences encoding the wild type or mutants of Hsp27. Then, these plasmids were respectively transfected along with psPAX2 and pMD2.G lentivirus packaging system plasmids (psPAX2 : pMD2.G : Plasmid of interest = 6 μg : 3 μg : 9 μg) into HEK293 T cells in 10-cm cell culture dishes using polyethylenimine (PEI). The medium was replaced after 2–4 h. After 48, 72, 96 h, the supernatant of cells was collected, aliquoted, and stocked at −80°C.
Immunofluorescence assay
We followed a reported protocol to study hnRNP A1 distribution [Citation18]. In brief, the cells on coverslips were infected with EV-A71 at the multiplicity of infection (MOI) of 40 for 6 h to assure the high efficiency of viral infection. Then the cells were fixed by 4% paraformaldehyde for 20 min, permeabilized by 0.5% Triton X-100 for 15 min, blocked with 5% BSA for 2 h, and stained by incubation with anti-Hsp27 (GTX101145) and anti-hnRNP A1 (sc-32301) antibody, then with Alexa Fluor 488 conjugated anti-rabbit antibody and Alexa Fluor 594 conjugated anti-mouse antibody respectively. After washing four times with 0.2% Triton X-100, the nuclei were stained with DAPI or Hoechst for 5 min. The images were captured by a Nikon A1HD25 High speed and Large Field of View Confocal Microscope. Colocalization analysis was conducted using the JACoP-plugin of the extended ImageJ version Fiji. The M1 colocalization coefficient was computed to illustrate the colocalization levels of Hsp27 and hnRNP A1 overlapped with Hoechst [Citation64].
Nuclear and cytoplasmic protein fraction
Nuclear and cytoplasmic protein was extracted using Beyotime Kit. The cells were washed once with PBS, scraped with a cell scraper, and collected by centrifugation. The supernatant was aspirated as hard as possible, leaving the cell pellet for later use. 200 µl of PMSF-supplemented Cytoplasmic Protein Extraction Reagent A per 20 µl of cell pellet was added to the cell pellet, which was vortexed vigorously at maximum speed for 5 s to completely suspend and disperse the cell pellet. The cell pellet was incubated on ice for 10–15 min. Then, 10 μl of Cytoplasmic Protein Extraction Reagent B was added to the mixture, which was vortexed vigorously at top speed for 5 s and incubated on ice for 1 min. After centrifuging at 16,000 xg for 5 min, the supernatant was collected in a pre-cooled plastic tube immediately, which is the cytoplasmic protein. For the pellet, the remaining supernatant was completely aspirated. 50 μl of PMSF-supplemented Nuclear Protein Extraction Reagent was added to the pellet. The mixture was vortexed vigorously at maximum speed for 15–30 s to completely suspend and disperse the cell pellet. Then the mixture was incubated on ice and vortexed at high speed for 15–30 s every 1–2 min for a total of 30 min. After centrifuging at 16,000 xg for 10 min at 4°C, the supernatant was collected in a pre-cooled plastic tube, which is the extracted nuclear protein.
Luciferase assay
Reporter plasmids were described previously [Citation43]. HEK 293 T cells were seeded in 24-wells, and then co-transfected with plasmids to simultaneously express 2Apro (200 ng), pIRES reporter (200 ng), and the wild-type Hsp27 or its mutants (800 ng). After culturing for 24 h, the cell lysates were collected using a passive lysis buffer (Promega, USA). The Renilla luciferase (RLuc) and Firefly luciferase (FLuc) activity were determined by using a dual-luciferase reporter assay (Promega) according to the manufacturer's instructions in a Lumat LB9507 bioluminometer.
Western blot assay
The protocol of western blot assay was described previously [Citation65]. After cell lysis in Nonidet-P40 (NP-40) buffer, the cell lysates were centrifuged, and the concentration was determined by Bradford assay (Bio-Rad). 20 μg of protein was used for 12% SDS-PAGE and transferred onto polyvinylidene fluoride membrane (PVDF) (GE, MA, USA). Following 5% bovine serum albumin (BSA) blocking for 1 h, the membrane was incubated with antibodies (1:1000) against Hsp27 (sc-13132), pHsp27-Ser15 (ab76313), pHsp27-Ser78 (ADI-SPA-523), pHsp27-Ser82 (ab155987), VP1 (GTX132339), β-Actin (sc-47778), hnRNP A1 (sc-32301), or GAPDH (sc-32233) overnight in 4°C. After secondary antibodies (HRP-conjugated Affinipure Goat Anti-Mouse IgG(H + L), SA00001-1, and HRP-conjugated Affinipure Goat Anti-Rabbit IgG(H + L), SA00001-2) incubation for 1 h, the protein was visualized with a chemiluminescence reagent (PerkinElmer, MA, USA) on Bio-Rad Chemidoc Imaging System. The protein bands were quantified by densitometry with ImageJ if necessary.
RNA isolation
RNA isolation was described before [Citation42]. The intracellular RNA was isolated with TRIzol reagent (Ambion, Life, Technologies), and then 1 μg total RNA was used to synthesize cDNA using PrimeScript™ RT Master Mix (Takara) according to the manufacturer's instructions.
Quantitative RT-PCR
Quantitative Real-time PCR (RT-qPCR) was performed using TB Green® Premix E × Taq™ (Takara) on Applied Biosystems QuantStudio™ 3 Real-Time PCR Systems. The target fragment amplification was carried out as follows: initial activation at 95°C for 30 s; PCR for 45 cycles: 95°C for 5 s, 60°C for 30 s. At the end of the amplification cycles, melting temperature analysis was carried out by a slow increase in temperature (0.1°C/s) up to 95°C. Primers of the target gene were EV-A71 VP1, F 5’-CGGACTGTAGGCACCTCGAA-3’, R 5’-CGCATTGGGCGAGGTATC-3’; GAPDH, F 5’-GTCTCCTCTGACTTCAACAGCG-3’, R 5’-ACCACCCTGTTGCTGTAGCCAA-3’. The messenger RNA (mRNA) level of each target gene was normalized to the mRNA copies of GAPDH in the same sample [Citation18].
Virus titration
RD cells were seeded into 96-well plates for 24 h, then cells were infected by 100 µl per well of serial 10-fold diluted supernatant in quintuplicate. The 50% tissue culture-infected dose (TCID50) was calculated by the Kärber method after 120 h of infection [Citation43,Citation52].
Peptide treatment
The peptides were constructed and purchased from GL Biochem (Shanghai) Ltd. RD cells were seeded into 24-well plates for 24 h, then cells were pre-treated with peptides at indicated concentrations for 2 h and infected by EV-A71 or transfected by plasmids for following experiments. The amino acids sequences of the peptides are RKKRRQRRR-AYSRALSRQLSS (TAT-S78, S78), RKKRRQRRR-AYSRALARQLSS (TAT-S78A, S78A), RKKRRQRRR-AYSRALSRQLAS (TAT-S82A, S82A), RKKRRQRRR-AYSRAAAAQLSS (TAT-S78-3A, S78-3A), RKKRRQRRR-YSREANQPKPSPKRESGEE (TAT-NC, NC).
CCK-8 assay
RD cells were seeded in 96-well cell plates (1 × 104/well) and placed in the 5% CO2 incubator for 1 day. Then the cells were treated with peptides for 2 days. 90 μl medium and 10 μl of CCK-8 (BioSharp, BS350B) was added to each well of the 96-well plate, which were incubated at 37°C for 1 h. A microplate reader (BioTek, Synergy H1) was used to measure the OD value of each well at 450 nm.
Statistical analysis
The results were expressed as mean ± standard deviation (SD). All statistical analysis was carried out with GraphPad 8.0 software (GraphPad Inc.). Two-tailed Student's t-test was applied for two-group comparison. A p value < 0.05 was considered statistically significant.
Abbreviation
Hsp27: heat shock protein 27; hnRNP A1: heterogeneous nuclear ribonucleoprotein A1; EV-A71: enterovirus A-71; 2Apro: 2A protease of enterovirus; 2AC110A: the protease-inactive mutation of 2Apro (Cys110 to Ala110); IRES: internal ribosome entry site; ssRNA: single-stranded RNA; HFMD: hand-foot-and-mouth disease; CNS: central nervous system; ITAFs: IRES trans-acting factors; MK2: MAPKAP kinases 2; sgRNA: single guide RNA; GFP: Green fluorescent protein; PEI: polyethylenimine; MOI: multiplicity of infection; RLuc: Renilla luciferase; FLuc: Firefly luciferase; TCID50: the 50% tissue culture-infected dose; WT: wild-type; Hsp27S15A, Hsp27S78A, and Hsp27S82A: the phosphorylation site of Hsp27 mutated from Serine to Alanine; Hsp27S15D, Hsp27S78D, and Hsp27S82D: the phosphorylation site of Hsp27 mutated from Serine to Asparticacid; Hsp27-3A: phosphorylation-deficient mutant Hsp27 S15/78/82A; Hsp27-3D: phosphorylation-active mimic Hsp27 S15/78/82D.
Conflicts of interest
The authors declare no conflict of interest.
Supplemental Material
Download MS Word (4.9 MB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Wan Q, Song D, Li H, et al. Stress proteins: the biological functions in virus infection, present and challenges for target-based antiviral drug development. Signal Transduct Target Ther. 2020;5(1):125. doi:10.1038/s41392-020-00233-4
- Tee HK, Zainol MI, Sam I-C, et al. Recent advances in the understanding of enterovirus A71 infection: a focus on neuropathogenesis. Expert Rev Anti Infect Ther. 2021;19(6):733–747. doi:10.1080/14787210.2021.1851194
- Liang L, Cheng Y, Li Y, et al. Long-term neurodevelopment outcomes of hand, foot and mouth disease inpatients infected with EV-A71 or CV-A16, a retrospective cohort study. Emerg Microbes Infect. 2021;10(1):545–554. doi:10.1080/22221751.2021.1901612
- Chang LY, Lin H-Y, Gau SS-F, et al. Enterovirus A71 neurologic complications and long-term sequelae. J Biomed Sci. 2019;26(1):57. doi:10.1186/s12929-019-0552-7
- Puenpa J, Wanlapakorn N, Vongpunsawad S, et al. The history of enterovirus A71 outbreaks and molecular epidemiology in the Asia-Pacific region. J Biomed Sci. 2019;26(1):75. doi:10.1186/s12929-019-0573-2
- Ng Q, He F, Kwang J. Recent progress towards novel EV71 anti-therapeutics and vaccines. Viruses. 2015;7(12):6441–6457. doi:10.3390/v7122949
- Zhang X, Zhang Y, Li H, et al. Hand-foot-and-mouth disease-associated enterovirus and the development of multivalent HFMD vaccines. Int J Mol Sci. 2022;24(1):169.
- Kok CC. Therapeutic and prevention strategies against human enterovirus 71 infection. World J Virol. 2015;4(2):78–95. doi:10.5501/wjv.v4.i2.78
- Tolbert M, Morgan CE, Pollum M, et al. HnRNP A1 alters the structure of a conserved enterovirus IRES domain to stimulate viral translation. J Mol Biol. 2017;429(19):2841–2858. doi:10.1016/j.jmb.2017.06.007
- Mahmud B, Horn CM, Tapprich WE. Structure of the 5’ untranslated region of enteroviral genomic RNA. J Virol. 2019;93(23):10–1128.
- Godet AC, David F, Hantelys F, et al. IRES trans-acting factors, key actors of the stress response. Int J Mol Sci. 2019;20(4):924. doi:10.3390/ijms20040924
- Sweeney TR, Abaeva IS, Pestova TV, et al. The mechanism of translation initiation on Type 1 picornavirus IRESs. EMBO J. 2014;33(1):76–92. doi:10.1002/embj.201386124
- Hung CT, Kung Y-A, Li M-L, et al. Additive promotion of viral internal ribosome entry site-mediated translation by far upstream element-binding protein 1 and an enterovirus 71-induced cleavage product. PLoS Pathog. 2016;12(10):e1005959. doi:10.1371/journal.ppat.1005959
- Lin JY, Li ML, Brewer G. mRNA decay factor AUF1 binds the internal ribosomal entry site of enterovirus 71 and inhibits virus replication. PLoS One. 2014;9(7):e103827. doi:10.1371/journal.pone.0103827
- Levengood JD, Tolbert M, Li M-L, et al. High-affinity interaction of hnRNP A1 with conserved RNA structural elements is required for translation and replication of enterovirus 71. RNA Biol. 2013;10(7):1136–1145. doi:10.4161/rna.25107
- Lin JY, Shih S-R, Pan M, et al. hnRNP A1 interacts with the 5’ untranslated regions of enterovirus 71 and Sindbis virus RNA and is required for viral replication. J Virol. 2009;83(12):6106–6114. doi:10.1128/JVI.02476-08
- De Jesus-Gonzalez LA, Palacios-Rápalo S, Reyes-Ruiz JM, et al. The nuclear pore complex is a key target of viral proteases to promote viral replication. Viruses. 2021;13(4):706. doi:10.3390/v13040706
- Dan X, Wan Q, Yi L, et al. Hsp27 responds to and facilitates enterovirus A71 replication by enhancing viral internal ribosome entry site-mediated translation. J Virol. 2019;93(9):10–1128.
- Singh MK, Sharma B, Tiwari PK. The small heat shock protein Hsp27: present understanding and future prospects. J Therm Biol. 2017;69:149–154. doi:10.1016/j.jtherbio.2017.06.004
- Tong SW, Yang Y-X, Hu H-D, et al. HSPB1 is an intracellular antiviral factor against hepatitis B virus. J Cell Biochem. 2013;114(1):162–173. doi:10.1002/jcb.24313
- Liu J, Zhang L, Zhu X, et al. Heat shock protein 27 is involved in PCV2 infection in PK-15 cells. Virus Res. 2014;189:235–242. doi:10.1016/j.virusres.2014.05.024
- Sreekanth GP, Chuncharunee A, Sirimontaporn A, et al. SB203580 modulates p38 MAPK signaling and dengue virus-induced liver injury by reducing MAPKAPK2, HSP27, and ATF2 phosphorylation. PLoS One. 2016;11(2):e0149486. doi:10.1371/journal.pone.0149486
- Kostenko S, Moens U. Heat shock protein 27 phosphorylation: kinases, phosphatases, functions and pathology. Cell Mol Life Sci. 2009;66(20):3289–3307. doi:10.1007/s00018-009-0086-3
- Xu L, Chen S, Bergan RC. MAPKAPK2 and HSP27 are downstream effectors of p38 MAP kinase-mediated matrix metalloproteinase type 2 activation and cell invasion in human prostate cancer. Oncogene. 2006;25(21):2987–2998. doi:10.1038/sj.onc.1209337
- Nayak TK, Mamidi P, Sahoo SS, et al. P38 and JNK mitogen-activated protein kinases interact with chikungunya virus non-structural protein-2 and regulate TNF induction during viral infection in macrophages. Front Immunol. 2019;10:786. doi:10.3389/fimmu.2019.00786
- Roy S, Roy S, Rana A, et al. The role of p38 MAPK pathway in p53 compromised state and telomere mediated DNA damage response. Mutat Res Genet Toxicol Environ Mutagen. 2018;836(Pt A):89–97. doi:10.1016/j.mrgentox.2018.05.018
- Peng H, Shi M, Zhang L, et al. Activation of JNK1/2 and p38 MAPK signaling pathways promotes enterovirus 71 infection in immature dendritic cells. BMC Microbiol. 2014;14:147. doi:10.1186/1471-2180-14-147
- Grimes JM, Grimes KV. p38 MAPK inhibition: a promising therapeutic approach for COVID-19. J Mol Cell Cardiol. 2020;144:63–65. doi:10.1016/j.yjmcc.2020.05.007
- Zhu S, Luo H, Liu H, et al. p38MAPK plays a critical role in induction of a pro-inflammatory phenotype of retinal Müller cells following Zika virus infection. Antiviral Res. 2017;145:70–81. doi:10.1016/j.antiviral.2017.07.012
- Fitzpatrick CJ, Mudhasani RR, Altamura LA, et al. Junin virus activates p38 MAPK and HSP27 upon entry. Front Cell Infect Microbiol. 2022;12:798978. doi:10.3389/fcimb.2022.798978
- Matsui T, Motoki Y, Inomoto T, et al. Temperature-related effects of adenosine triphosphate-activated microglia on pro-inflammatory factors. Neurocrit Care. 2012;17(2):293–300. doi:10.1007/s12028-011-9639-z
- Guo K, Liu Y, Zhou H, et al. Involvement of protein kinase C beta-extracellular signal-regulating kinase 1/2/p38 mitogen-activated protein kinase-heat shock protein 27 activation in hepatocellular carcinoma cell motility and invasion. Cancer Sci. 2008;99(3):486–496. doi:10.1111/j.1349-7006.2007.00702.x
- van de Klundert FA, Gijsen MLJ, van den IJssel PRLA, et al. alpha B-crystallin and hsp25 in neonatal cardiac cells–differences in cellular localization under stress conditions. Eur J Cell Biol. 1998;75(1):38–45. doi:10.1016/S0171-9335(98)80044-7
- Lavoie JN, Gingras-Breton G, Tanguay RM, et al. Induction of Chinese hamster HSP27 gene expression in mouse cells confers resistance to heat shock. HSP27 stabilization of the microfilament organization. J Biol Chem. 1993;268(5):3420–3429. doi:10.1016/S0021-9258(18)53711-X
- Arrigo AP, Suhan JP, Welch WJ. Dynamic changes in the structure and intracellular locale of the mammalian low-molecular-weight heat shock protein. Mol Cell Biol. 1988;8(12):5059–5071.
- Mathew SS, Della Selva MP, Burch AD. Modification and reorganization of the cytoprotective cellular chaperone Hsp27 during herpes simplex virus type 1 infection. J Virol. 2009;83(18):9304–9312. doi:10.1128/JVI.01826-08
- Trott D, McManus CA, Martin JL, et al. Effect of phosphorylated hsp27 on proliferation of human endothelial and smooth muscle cells. Proteomics. 2009;9(12):3383–3394. doi:10.1002/pmic.200800961
- Li H, Wang X, Wang Y, et al. Secreted LRPAP1 binds and triggers IFNAR1 degradation to facilitate virus evasion from cellular innate immunity. Signal Transduct Target Ther. 2023;8(1):374. doi:10.1038/s41392-023-01630-1
- Reyes-Del Valle J, Chávez-Salinas S, Medina F, et al. Heat shock protein 90 and heat shock protein 70 are components of dengue virus receptor complex in human cells. J Virol. 2005;79(8):4557–4567. doi:10.1128/JVI.79.8.4557-4567.2005
- Tsou YL, Lin Y-W, Chang H-W, et al. Heat shock protein 90: role in enterovirus 71 entry and assembly and potential target for therapy. PLoS One. 2013;8(10):e77133. doi:10.1371/journal.pone.0077133
- Ujino S, Yamaguchi S, Shimotohno K, et al. Heat-shock protein 90 is essential for stabilization of the hepatitis C virus nonstructural protein NS3. J Biol Chem. 2009;284(11):6841–6846. doi:10.1074/jbc.M806452200
- Zhou F, Wan Q, Lu J, et al. Pim1 impacts enterovirus A71 replication and represents a potential target in antiviral therapy. iScience. 2019;19:715–727. doi:10.1016/j.isci.2019.08.008
- Dong Q, Men R, Dan X, et al. Hsc70 regulates the IRES activity and serves as an antiviral target of enterovirus A71 infection. Antiviral Res. 2018;150:39–46. doi:10.1016/j.antiviral.2017.11.020
- Ma Y, Yu J, Chan HLY, et al. Glucose-regulated protein 78 is an intracellular antiviral factor against hepatitis B virus. Mol Cell Proteomics. 2009;8(11):2582–2594. doi:10.1074/mcp.M900180-MCP200
- Karaca G, Hargett D, McLean TI, et al. Inhibition of the stress-activated kinase, p38, does not affect the virus transcriptional program of herpes simplex virus type 1. Virology. 2004;329(1):142–156. doi:10.1016/j.virol.2004.08.020
- Fukagawa Y, Nishikawa J, Kuramitsu Y, et al. Epstein-Barr virus upregulates phosphorylated heat shock protein 27kDa in carcinoma cells using the phosphoinositide 3-kinase/Akt pathway. Electrophoresis. 2008;29(15):3192–3200. doi:10.1002/elps.200800086
- Song C, Liu H, Cao Z, et al. HSP27 interacts with nonstructural proteins of porcine reproductive and respiratory syndrome virus and promotes viral replication. Pathogens. 2023;12(1):91. doi:10.3390/pathogens12010091
- Nakatsue T, Katoh I, Nakamura S, et al. Acute infection of Sindbis virus induces phosphorylation and intracellular translocation of small heat shock protein HSP27 and activation of p38 MAP kinase signaling pathway. Biochem Biophys Res Commun. 1998;253(1):59–64. doi:10.1006/bbrc.1998.9724
- Fan S, Xu Z, Liu P, et al. Enterovirus 71 2A protease inhibits p-body formation to promote viral RNA synthesis. J Virol. 2021;95(19):e0092221.
- Yang X, Hu Z, Fan S, et al. Picornavirus 2A protease regulates stress granule formation to facilitate viral translation. PLoS Pathog. 2018;14(2):e1006901. doi:10.1371/journal.ppat.1006901
- Tang WF, Huang R-T, Chien K-Y, et al. Host microRNA miR-197 plays a negative regulatory role in the enterovirus 71 infectious cycle by targeting the RAN protein. J Virol. 2016;90(3):1424–1438. doi:10.1128/JVI.02143-15
- Lu J, Yi L, Zhao J, et al. Enterovirus 71 disrupts interferon signaling by reducing the level of interferon receptor 1. J Virol. 2012;86(7):3767–3776. doi:10.1128/JVI.06687-11
- Zheng W, Zhou Z, Rui Y, et al. TRAF3 activates STING-mediated suppression of EV-A71 and target of viral evasion. Signal Transduct Target Ther. 2023;8(1):79. doi:10.1038/s41392-022-01287-2
- Lizcano-Perret B, Michiels T. Nucleocytoplasmic trafficking perturbation induced by picornaviruses. Viruses. 2021;13(7):1210. doi:10.3390/v13071210
- Zhang G, Zhou F, Gu B, et al. In vitro and in vivo evaluation of ribavirin and pleconaril antiviral activity against enterovirus 71 infection. Arch Virol. 2012;157(4):669–679. doi:10.1007/s00705-011-1222-6
- Tijsma A, Franco D, Tucker S, et al. The capsid binder Vapendavir and the novel protease inhibitor SG85 inhibit enterovirus 71 replication. Antimicrob Agents Chemother. 2014;58(11):6990–6992. doi:10.1128/AAC.03328-14
- Senior K. FDA panel rejects common cold treatment. Lancet Infect Dis. 2002;2(5):264. doi:10.1016/S1473-3099(02)00277-3
- Egorova A, Ekins S, Schmidtke M, et al. Back to the future: advances in development of broad-spectrum capsid-binding inhibitors of enteroviruses. Eur J Med Chem. 2019;178:606–622. doi:10.1016/j.ejmech.2019.06.008
- Lalani S, Gew LT, Poh CL. Antiviral peptides against Enterovirus A71 causing hand, foot and mouth disease. Peptides. 2021;136:170443. doi:10.1016/j.peptides.2020.170443
- Wang Z, Yi B, Wu M, et al. Bioinspired supramolecular slippery organogels for controlling pathogen spread by respiratory droplets. Adv Funct Mater. 2021;31(34):2102888. doi:10.1002/adfm.202102888
- Hou C, Chang Y-F, Yao X. Supramolecular adhesive materials with antimicrobial activity for emerging biomedical applications. Pharmaceutics. 2022;14(8):1616. doi:10.3390/pharmaceutics14081616
- Yi L, He Y, Chen Y, et al. Potent inhibition of human enterovirus 71 replication by type I interferon subtypes. Antivir Ther. 2011;16(1):51–58. doi:10.3851/IMP1720
- Li C, Huang L, Sun W, et al. Saikosaponin D suppresses enterovirus A71 infection by inhibiting autophagy. Signal Transduct Target Ther. 2019;4:4. doi:10.1038/s41392-019-0037-x
- Zinchuk V, Zinchuk O, Okada T. Quantitative colocalization analysis of multicolor confocal immunofluorescence microscopy images: pushing pixels to explore biological phenomena. Acta Histochem Cytochem. 2007;40(4):101–111. doi:10.1267/ahc.07002
- Ma Y, Peng J, Liu W, et al. Proteomics identification of desmin as a potential oncofetal diagnostic and prognostic biomarker in colorectal cancer. Mol Cell Proteomics. 2009;8(8):1878–1890. doi:10.1074/mcp.M800541-MCP200