
ABSTRACT
The global outbreak of Mpox, caused by the monkeypox virus (MPXV), has attracted international attention and become another major infectious disease event after COVID-19. The mRNA cap N7 methyltransferase (RNMT) of MPXV methylates the N7 position of the added guanosine to the 5'-cap structure of mRNAs and plays a vital role in evading host antiviral immunity. MPXV RNMT is composed of the large subunit E1 and the small subunit E12. How E1 and E12 of MPXV assembly remains unclear. Here, we report the crystal structures of E12, the MTase domain of E1 with E12 (E1CTD-E12) complex, and the E1CTD-E12-SAM ternary complex, revealing the detailed conformations of critical residues and the structural changes upon E12 binding to E1. Functional studies suggest that E1CTD N-terminal extension (Asp545-Arg562) and the small subunit E12 play an essential role in the binding process of SAM. Structural comparison of the AlphaFold2-predicted E1, E1CTD-E12 complex, and the homologous D1-D12 complex of vaccinia virus (VACV) indicates an allosteric activating effect of E1 in MPXV. Our findings provide the structural basis for the MTase activity stimulation of the E1-E12 complex and suggest a potential interface for screening the anti-poxvirus inhibitors.
Introduction
Monkeypox virus (MPXV) is classified as an orthopoxvirus within the poxviridae family, exhibiting the capacity to induce zoonotic viral diseases. Human cases of MPXV were initially documented sporadically in Africa during the 1970s [Citation1]. Since 2022, there has been a dramatic increase in the number of MPXV cases globally, with the number of detected MPXV infections soaring to more than 48,000 and a total of 13 deaths recorded in less than four months [Citation2]. This epidemiological upswing prompted the World Health Organization (WHO) to officially declare the monkeypox outbreak a global health emergency [Citation3]. MPXV is one of the largest and most complex double-stranded DNA viruses, showing a brick-shaped structure with a length of 220–450 nm and a width range of 140–260 nm. It has four components: the core, the lateral bodies, the outer membrane, and the outer lipoprotein envelope, of which the core is the central part surrounded by core fibrils and double-stranded viral DNA [Citation4].
During poxvirus-host coevolution, viral mRNA has the same cap structure as host mRNA, which allows the virus to evade detection by the innate immune system and produce viral proteins efficiently [Citation5]. The synthetic cap structure consists of three typical capping reactions [Citation6]. First, RNA triphosphatase (RTPase) hydrolyses γ-phosphate (pppNp-RNA) of nascent RNA to generate diphosphate RNA (ppNp-RNA) and inorganic phosphate (Pi). The guanyltransferase (GTase) then reacts with the α-phosphate of GTP, releasing pyrophosphate (PPi) and forming a covalent enzyme-guanylate intermediate (Gp-GTase). GTase then transfers the GMP molecule (Gp) into the 5 ‘-diphosphate RNA to produce GpppNp-RNA [Citation7]. In the final step, guanine-N7-methyltransferase (N7MTase) transfers methyl groups from S-adenosine-L-methionine (SAM or AdoMet, which can provide methyl groups to methylate the substrate) to coronoguanine to form the cap-0 structure, 7-methyl-GPPPNP (m7GpppNp), and releases S-adenosine-L-homocysteine (SAH or AdoHcy) as a by-product [Citation7].
MPXV belongs to the poxvirus family and encodes a heterodimeric RNA capping enzyme consisting of a large subunit E1 and a small subunit E12 [Citation8]. The RNA capping enzymes encoded by poxviruses share a high degree of homology among the poxvirus family. The E1 subunit of the MPXV contains catalytic modules required to produce cap 0, as does the D1 subunit of the vaccinia virus [Citation8]. The catalytic modules are neatly arranged in the order of the N-terminal RTPase, the middle GTase, and the C-terminal domain (CTD) composed of guanine-N7 MTase [Citation9]. The guanine-N7 MT enzyme activity of the vaccinia virus requires an association with D12 to function efficiently [Citation10]. Considering the high homology between E12 and D12, it is postulated that the CTD of E1 (denoted as E1CTD) forms a stable complex with E12. Reports showed that the interaction of D12 with the guanine-N7 MTase domain of vaccinia virus capping enzymes induces the conformational changes required for efficient catalysis [Citation11]. However, how the conformational change occurs and the transition from GTase to MTase active site in MPXV remains unclear.
In this study, we solved the MPXV E12, the E1CTD-E12 complex, and the E1CTD-E12-SAM ternary complex structures using X-ray crystallography. E1CTD and E12 form a 1:1 heterodimeric complex in solution and crystal structure. Mutations in the E1CTD – E12 interface dramatically decreased the binding affinity. Structural comparison of E12 and E1CTD-E12 complex showed significant conformation changes upon E12 binding to E1. In addition, we found the E1CTD N-terminal extension (Asp545-Arg562) and the small subunit E12 play an essential role in SAM binding. Superimposition of the AlphaFold2-predicted structure of full-length E1 with different homologs revealed that bound E12 could stimulate E1 activity and form a compact complex. This may provide the structural basis for the MTase activity stimulation of the E1-E12 complex and suggest a potential interface for screening the anti-poxvirus inhibitors.
Results
The overall structure of the MPXV E1CTD-E12 MTase complex
To reveal the molecular basis of the MPXV MTase complex in evading host antiviral immunity, we investigated the crystal structure of the E1-E12 complex. The full-length MPXV E1 contains three domains, named RTPase domain (residues 1-225), GPase domain (residues 226-502), and N7 MTase domain (CTD, residues 545-844) (A). E1 interacts with E12 to form the MPXV MTase complex via the N7 MTase domain (E1CTD-E12). As obtaining the full-length E1-E12 complex is challenging, we successfully co-expressed and purified the E1CTD-E12 complex in E. coli. The E1CTD-E12 complex came out at the elution peak of 79 mL on a Superdex200 16/600 GL column, corresponding to a molecular mass of 68 kDa (B).
Figure 1. Crystal structure of the MPXV E1CTD-E12 complex. (A) The domain architectures of E1 and E12 protein. The RTPase, GPase, and N7 MTase domains (denoted as E1CTD) of MPXV E1 are coloured in gray, blue, and orange, respectively. E12 is coloured in green. (B) Size-exclusion chromatographic profile and SDS-PAGE result of the E1CTD-E12 complex on a Superdex200 16/600 GL column. M: protein marker. (C) A ribbon representation of the E1CTD-E12 complex structure, showing 14 α-helices and 23 β-sheets. The E1CTD and E12 are coloured in orange and green, respectively. (D) The electrostatic potential surfaces of the E1CTD-E12 complex. Red indicates negative potential, and blue indicates positive potential.
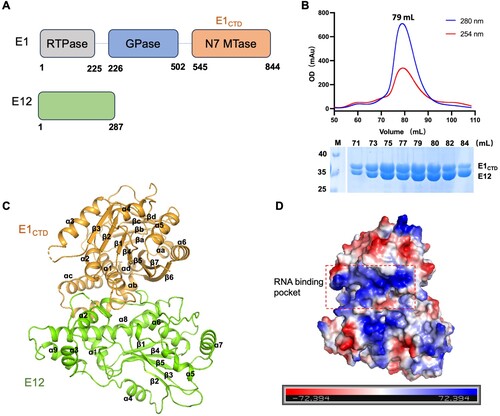
After several rounds’ optimization of crystallization conditions, we successfully solved the E1CTD-E12 crystals structure, which belonged to the space group P6522 at a resolution of 3.94 Å (C and ). There is one molecule of E1CTD-E12 within the asymmetric unit, composed of 14 α-helices and 23 β-sheets (C). The electrostatic potential surfaces of the E1CTD-E12 complex are evenly distributed with positive and negative charges. However, the RNA-binding pocket of E1, adjacent to E12, was full of positive charge (D).
Table 1. Data collection and refinement statistics.
The interface between E1CTD and E12
Previous studies showed that the MTase domain of VACV D1 has a feeble intrinsic biochemical activity and requires a stimulating subunit (D12) to exert full MTase activity. The MPXV capping enzyme has a C-terminal MTase domain of E1 subunit and a stimulatory subunit E12. The structure of E1CTD comprised 11 β-strands and 10 α-helices. The overall view indicates that E1CTD has a classic SAM-MT fold [Citation12], incorporated seven alternating β-strands, namely β1 (Lys593-Ile597), β2 (Leu615-Thr619), β3 (Lys646-Ile650), β4 (Ile674-Gln678), β5 (Lys706-Met712), β6 (Phe780-Asp787), and β7 (Tyr836-Lys843), surrounded by six α-helices, namely, α1 (Gly563-Tyr580), α2 (Leu605-Gly611), α3 (Asp624-Leu636), α4 (Asp657-Glu665), α5 (Tyr690-Ser698), and α6 (Lys768-Glu777). Different from the core SAM-MT fold, E1CTD has one α-helix (αa) and four alternating β-strands (βa, βb, βc, βd) between β5 and α5, and three α-helices (αb, αc, αd) between β6 and β7 (Figures S1 and 2A).
The E12 subunit exhibits a high affinity to the E1CTD domain. The interface-involved residues of E12 were located in the α2 helix (Leu34-Arg49), while the related residues of E1CTD were mainly located in regions Tyr571-Asp587 (α1) and Glu806-Phe814 (αb-αc) (A). Electrostatic and hydrophobic analysis showed that the inner part of the interface is mainly hydrophobic, while the ends are electrostatic interaction. Briefly, residues Lys31 and Arg49 on the E12 formed salt bridges with Asp587 and Glu814 on the E1CTD, respectively. Also, there is extensive hydrophobic interaction at the E1CTD-E12 interface. Residues Leu37 and Phe44 on the E12 formed hydrophobic interaction with Leu575 and Phe814 on the E1CTD, and the side chain of E12 Leu34 formed hydrophobic interaction with the side chain of E1CTD Leu586 residue (B). Next, we investigated the interaction between E1CTD and E12 using the isothermal titration calorimetry (ITC) and surface plasmon resonance (SPR) methods. SPR result showed a KD value of 8.15 nM and ITC result showed a KD value of 35.5 nM (C and D), indicating that the E12 subunit exhibits a high affinity to the E1CTD domain.
Figure 2. MPXV E12 exhibits a high binding affinity with E1CTD. (A) Cartoon representation of MPXV E1CTD-E12 complex structure, the binding interface was labelled with a red rectangle for key residues depicted with stick models. (B) Ribbon representations of crucial residues in the interface between E12 (green) and E1CTD (orange). The residues were labelled and shown as sticks. The hydrogen bonds among interacted residues are shown in yellow dashed lines. (C) Measurements of the binding affinity between E1CTD and E12 by ITC method. The upper panel represents the raw data, revealing the calorimetric response peaks during successive injections of E12 to the E1CTD plot. The lower panel is the best fit of the raw data after subtracting the heat of dilution from the appropriate buffer blank. The solid line in the lower panel shows the best fit of the raw data to the one-set of sites binding model. (D) SPR assay showing the binding of E1CTD and E12. (E) The gel-filtration profiles of the E1CTD-E12 complex, E12, and different mutants. (F) The SDS-PAGE results of the E1CTD-E12 complex, E12, and different mutants, corresponding to (E). M: protein marker.
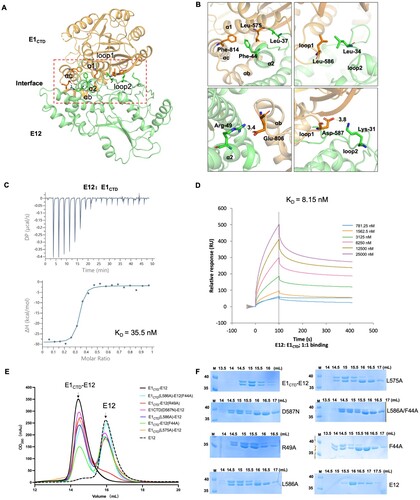
To identify the critical residues for the interaction of the E1CTD-E12 complex, we constructed six E1CTD-E12 mutants, including F44A and R49A in E12, D587N, L575A, L586A, and double mutant F44A/L586A in E1CTD-E12. The size exclusion chromatography results of E1CTD-E12 mutants clearly showed that F44A, D587N, L575A, L586A, and double mutant F44A/L586A significantly decreased the binding of the E1CTD and E12. However, these mutants did not destroy the E1CTD-E12 formation, indicating that the interaction of E1CTD and E12 is coordinated by a variety of amino acids (E and F).
The solution structures of E12 and E1CTD-E12 complex
To reveal how E12 bound and stimulated E1 activity, we expressed and purified E12 protein in E. Coli. The E12 was eluted out at the peaks of 83.5 mL on the gel-filtration column, corresponding to a mass of 33 kDa (Figure S2A). Next, the E12 structure was solved by the X-ray crystallography method with a resolution of 2.2 Å in the P3121 space group (). The asymmetric unit of the crystal structure consisted of one E12 molecule. A central feature of the E12 is a strip of parallel and anti-parallel β-strands (β1, β4, β5, β3, β2), which is stabilized by surrounding nine α-helices (A). In order to explore whether there will be structural changes in E1 upon binding with E12, we superimposed the E12 to the E1CTD-E12 structures and found that the overall folding of E12 is very similar in the structures; the root mean square deviation (RMSD) value is 0.544 Å, based on 253 pairs of Cα atoms (B). The conformations of the α-helices and β-strands are well conserved, but E12 in the E1CTD-E12 does show some conformational changes, especially between the Lys118 and Asn126. The density of Asn120 to Glu125 was not observed in any reported E1CTD-E12 structures, which might be due to the side chain of His122 being too close to the side chain of Lys583 in E1CTD (B). We tried to construct mutants E1CTD-E12 (H122D) and E1CTD-E12 (K583A) to stabilize the E1CTD-E12 complex structure and carried out crystallization trials. Fortunately, the E1CTD-E12 (H122D)-SAM structure was solved by the X-ray crystallography method with a resolution of 3.3 Å in the P6522 space group () and the small molecule SAM is clearly observed in the structure.
Figure 3. The solution structures of E12 and E1CTD-E12 complex. (A) Cartoon presentation of the E12 structure, showing nine α-helices and five β-sheets. (B) Structural superimposition of the E1CTD-E12 (orange and green) and E12 (purple). Residues His122 of E12 and Lys583 of E1CTD are shown in stick models. (C) The experimental scattering curve of the MPXV E1CTD-E12 complex. (D) The distance distribution function curve of the E1CTD-E12 complex. (E) The crystal structure of the E1CTD-E12 complex was fitted into the ab initio envelope obtained from SAXS. (F) The experimental scattering curve of MPXV E12. (G) The distance distribution function curve of E12. (H) The crystal structure of E12 was fitted into the ab initio envelope obtained from SAXS.
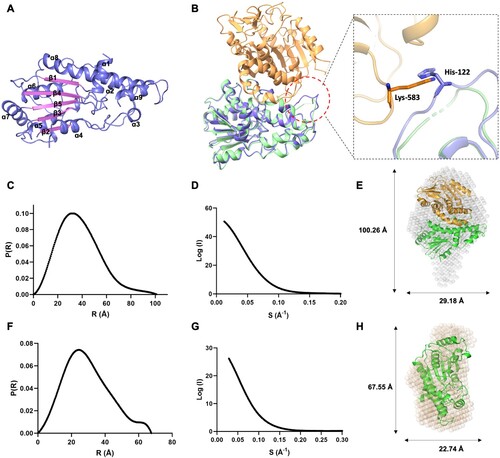
In addition, we investigated the solution status of the E12 and E1CTD-E12 complex by the small-angle X-ray scattering (SAXS) and DLS methods (C-H and Table S1). All the proteins behaved well in the solution. The maximum dimension (Dmax) from distance distribution function p(r) for the E12 and E1CTD-E12 complex were 67.5 and 100.4 Å, respectively (C and F). When superimposed the E12 and E1CTD-E12 complex crystal structures with the envelopes generated from SAXS data, the E12 and E1CTD-E12 complex showed high similarities (E and H).
Analysis of the E1CTD-SAM binding model
To elucidate the molecular basis of MPXV mRNA cap N7 methyltransferase, we successfully obtained the E1CTD-E12 (H122D)-SAM ternary complex structure. The SAM molecule is bound in the pocket consisting of four loops (NTD-α1, β1-α1, β2-α3, and α4-α5), forming a closed substrate binding site (A). The directly interacting amino acids surrounding the SAM-binding region include Arg548, Leu549, Asn550, Lys573, Asp598, Asp620, Arg655, Gln678, and Tyr683. The adenosine moiety is stacked between conserved Tyr683 and Arg548. In addition, the main chain of Leu549 and Asn550 forms hydrogen bond (H-bond) interaction with the adenine N6 and N1, respectively, and the side chain of Asn550 forms H-bond interaction with the adenine N7. The O atom of the Gln678 and Asp598 forms an H-bond with the homocysteine of the AdoMet. Moreover, the – COOH of Asp620 and the – NH3 of Lys573 form polar contacts with – OH of the adenosine ribose and – COOH in the SAM, respectively (A).
Figure 4. Comparison of the SAM binding pocket in MPXV mRNA capping MTase and other homologs. (A) The overall structure of the E1CTD-E12 (H122D)-SAM ternary complex in cartoon presentation. The zoomed-in area shows the detailed contact analysis between SAM (cyan sticks) and MPXV E1CTD (orange cartoon). The substrate SAM is shown in a stick model with an electron density map (2Fo-Fc, level = 1.0). (B) Structure comparison of MPXV mRNA capping MT E1CTD (orange, PDB: 8ZE4) with VACV D1CTD (pink, PDB: 2VDW). The adenosine nucleoside of SAM in E1 and SAH in D1 adopts a syn-conformation, respectively. (C) Structure comparison of RNA capping MTase members from MPXV (orange, PDB: 8ZE4), African Swine Fever Virus (ASFV, blue, PDB: 7D8U), and Encephalitozoon cuniculi (Ecm1, gray, PDB: 1RI3). The adenosine nucleoside of SAM in E1 (orange) of MPXV adopts a syn-conformation, whereas those in ASFV (blue) and Ecm1 (gray) adopt an anti-conformation. (D) Multiple sequence alignment of MPXV E1 with VACV D1 (PDB: 2VDW), pNP868RMT of ASFV (PDB: 7D8U), RMNT of Homo sapiens (PDB: 3BGV), and ABD1 of Ecm1 (PDB: 1RI3). Identical and similar residues among groups are shown in white font on a red background and in red font on a white background, respectively.
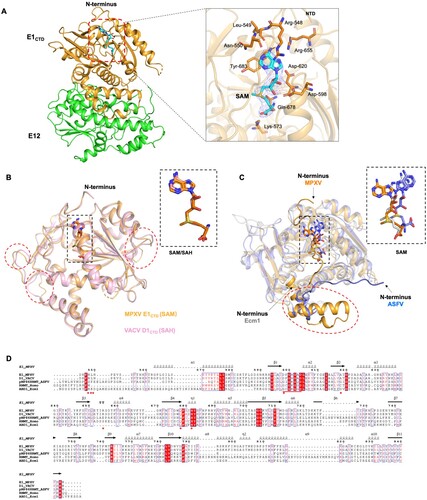
Superimposition of the MPXV E1CTD-E12 (H122D)-SAM complex structure with the VACV D1CTD-D12 complex (PDB ID: 2VDW) [Citation11], we found poxvirus mRNA cap N7 methyltransferase are highly conserved in overall structures. The RMSD values between the E1CTD-E12 and D1CTD-D12 complexes were 0.430 Å among 276 Cα atoms, only showing some differences in the α linker and β linker regions (B). Structure superimposing the MPXV E1CTD-SAM complex with that of Encephalitozoon (PDB ID: 1RI3) and African swine fever virus ASFV (PDB ID: 7D8U) revealed similar features of core SAM-MT fold, whereas the additions to the core fold are quite differences (C). The significant difference was that the MPXV E1CTD had the N-terminal extension (Asp545-Arg562) before the α1 helix. The N-terminal peptide of VACV D1CTD involved in SAM binding (closed conformation) and protected D1CTD from trypsinization [Citation11]. Another minor difference was that the MPXV E1CTD had three helices αb (Phe788-Met805), αc (Pro809 – Lys823), αd (Val829 – Tyr835) between β6 and β7. The corresponding regions are involved in residues interacting with E12 to help form a stable E1CTD-E12 complex structure (C).
In addition, we compared the catalytic sites of E1CTD-E12 with the catalytic sites of MTases from unrelated eukaryotes, prokaryotes, and viruses, namely Homo sapiens, Encephalitozoon, ASFV, and Mimivirus [Citation13–15]. The amino acid sequences alignment of capping enzyme MTase domain found MTases from Homo sapiens, Encephalitozoon, ASFV and Mimivirus have a conserved SAM-binding motif (Gln/Asp-X-Gly-X-Gly-X-Gly). In contrast, the sequence of the poxvirus's SAM-binding domain (Ala-X-Asp-X-Gly-X-Gly) is slightly different (D) [Citation12]. Previous structural studies of VACV MTase showed that Asp598 is substituted for the usually highly conserved glycine, making the adenosine adopt the usual syn-conformation in the active site [Citation11]. Also, the essential amino acids that interacted with SAM in poxviruses are not exactly conserved in other MTases (D).
Structural basis for the allosteric effect of capping enzyme in poxviruses
We then performed the interaction assay between E1CTD-E12 and SAM by ITC. The KD value of 3.87 μM was determined using the one-site binding site model (A). The mutated complex Δ(545-562) E1CTD-E12 (deletion of Asp545-Arg562) shows no detectable binding affinity to SAM (B), suggesting that the N-terminal extension (Asp545-Arg562) plays a vital role in the substrate binding process, consisting with the observation in the complex structure (A). In addition, the E1CTD also shows no detectable binding ability to SAM (C), indicating that the binding of the small subunit E12 may lead to structure changes in the large subunit E1CTD.
Figure 5. Binding characteristics of MPXV mRNA capping MTase with its substrate SAM. (A-C) The binding affinity of SAM with E1CTD-E12 complex (A), Δ(545-562) E1CTD-E12 complex (B), and E1CTD (C), respectively. Baseline-subtracted raw ITC data for injections of SAM are indicated in the upper panels of each ITC profile shown. The peaks normalized to the ligand/protein molar ratio were integrated, as shown in the bottom panels. The solid line in the lower panel shows the best fit of the raw data to the one-set of sites binding model. (D) Structural comparison of AlphaFold2-predicted E1 (wheat) with solved E1CTD-E12 complex (green, PDB: 8Y2Z), shown in wheat and green, respectively. (E) Structural comparison of AlphaFold2-predicted E1 (wheat) with VACV D1-D12 complex (cyan, PDB: 4CKC), shown in wheat and cyan, respectively. The red arrow indicates the rotation of the MTase domain upon binding with E12.
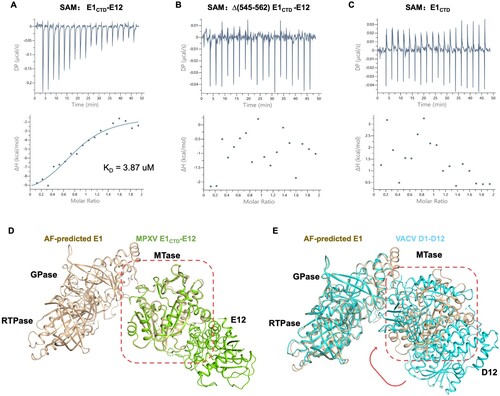
Next, we compared the structure of E1CTD-E12 with the structure of E1 predicted by AlphaFold2. Structural superposition showed that the AlphaFold2-predicted MTase domain structure in full-length E1 is very similar to the resolved E1CTD-E12 structure, with an RMSD value of 0.451 Å among 269 Cα atoms (D). Interestingly, when superimposition the AlphaFold2-predicted E1 with the VACV D1-D12 complex (PDB ID: 4CKC), the N7 MTase domain is found to be close to the GTase domain with an RMSD value of 0.667 Å among 514 Cα atoms (E). The amino acid residues (Ile528-Asn544) at the junction of MTase and GTase were not observed in the D1-D12 complex structure, which might be due to this region being more flexible. Previous studies indicated that poxviruses encode a small subunit that stimulates MTase activity. In the Vaccinia virus, the small subunit D12 is essential; it lacks any MT activity but can stimulate the weak intrinsic MT activity of D1 by 30–50 folds [Citation16]. The previous study and our alignment results confirmed that the binding of the small subunit E12 of the MPXV capping enzyme may lead to an allosteric reaction of the MTase in the large subunit E1 [Citation11]. This reaction makes the overall structure more compact, and the substrate binding sites in the GTase and MTase domains are closer, which may be more conducive to the enzymatic reaction and the transfer of substrate in different subunits.
Discussion
The mRNA cap structure was discovered in the 1970s, and numerous studies have led to a deeper understanding of the biological role of mRNA cap structures. The mRNA cap structure plays an important role in protein translation and innate immunity [Citation17]. Therefore, the virus can produce its own mRNA cap structure, which can effectively synthesize viral proteins and evade the innate immune response of host cells [Citation18]. Due to the wide variety of viruses, mRNA capping has various mechanisms. The GTase and RTPase activities of Chlorella virus (dsDNA) are located in different proteins [Citation19]. Vaccinia virus (dsDNA) and Bluetongue virus (dsRNA) encode multifunctional proteins that produce cap 0 and cap 1 RNA, respectively [Citation20]. Sars-cov-2 ((+) ssRNA) can use the single-stranded nucleic acid binding protein (nsp9) in its transcription and replication complex as a medium to mediate the novel capping process [Citation21]. Flaviviruses ((+) ssRNA) can tether the activities of GTase and MTase to their RNA-dependent RNA polymerase (RdRp) [Citation22]. Alphaviruses ((+) ssRNA) reorder the enzymatic steps of RNA capping. GTP is first modified to become m7GMP and then transferred to processed ppRNA to form m7G-cap [Citation23]. Currently, only the full-length protein structure of vaccinia virus capping enzymes has been fully resolved. In the structure, GTP and SAH identify the GTase and MTase active sites [Citation9]. However, how the substrate moves from one active site to the next and whether conformational changes occur during these transitions have not been observed in any crystalline structure.
We solved the MPXV MT subcomplex E1CTD-E12 crystal structures and the small subunit E12. By analyzing the interaction interface between E1 and E12 in E1CTD-E12, it was found that the large and small subunits interacted with each other mainly through hydrophobic interaction ( and ). In the binding process between E12 and E1, the loop of small subunit E12 conformationally changed due to the steric hindrance between the side chain of His122 and Lys583 (B). The substrate binding sites in the domain of the MPXV MTase were found to be remarkably conserved by structural comparison (A and B). However, there were still significant differences between the poxvirus MTases and other MTases. The first is amino acid residues change in the conserved SAM binding site (D). Second is the presence of the small subunit E12 that can stimulate E1-MTase activity, which is also unique to poxviruses. The ITC result of Δ(545-562) E1CTD-E12 and E1CTD with SAM suggest the essential role of the N-terminal extension (Asp545-Arg562) and the small subunit E12 in substrate binding process (A-C). Despite the high homology between VACV D1CTD-D12 and MPXV E1CTD-E12, the mutation study showed that the residue Tyr202 of MPXV (corresponding to residue Cys202 in VACV) can dramatically change the complex homogeneity in gel-filtration profiles (Figure S2C). Moreover, we predicted the full-length E1 protein with the help of AlphaFold2 and found that the structure of the full-length E1 protein was significantly different from that of the full-length protein bound to the small subunit. The differences mainly occurred in the MTase domain with a large allosteric reaction (D and E). We speculate that the binding of small subunits may make the structure of large subunits more compact, bringing the active sites of different domains closer together, which is more conducive to the movement of substrates between active sites and the occurrence of enzymatic active reactions (D and E). Overall, the structure of poxvirus capping enzymes is highly conserved, suggesting an essential role in the viral life cycle. Therefore, it is promising to use MPXV capping enzymes as targets for anti-poxvirus drug discovery.
Methods and materials
Plasmid construction
The full-length E12 (UniProt ID: Q8V4X6) gene and the full-length E1 (UniProt ID: Q8V4Y7) were synthesized by Tsingke, the target genes were amplified by the polymerase chain reaction and subcloned into the pSMT3 vector with a His-SUMO tag. The E1CTD (residues 545-844) were subcloned into the pSMT3 vector with a His-SUMO tag. The MTase subcomplex E1CTD-E12 and Δ(545-562) E1CTD-E12 were subcloned into the pETDuet vector with a 6xHis tag. The E1CTD-E12 mutants were generated by whole-plasmid PCR in an 18-cycle reaction with steps at 95 °C for 15 s, 65 °C for 30 s, and 72 °C for 3 min per cycle. After digestion with the enzyme DpnI, the PCR products were transformed into E. coli DH5a cells. The positive constructs were determined by DNA sequencing. Mutants F44A, R49A, H122D, Y202C and Y202A in E12, K583A, D587N, L575A, L586A, and double mutant F44A/L586A in E1CTD-E12 were constructed according to the standard QuikChange Site-Directed Mutagenesis protocol (Stratagene, United States), confirmed by DNA sequencing finally.
Protein expression and purification
The wild-type and mutants of E12, E1CTD, E1CTD-E12, and Δ(545-562) E1CTD-E12 were transformed into E. coli BL21 (DE3) cells for protein expression. E. coli cells were cultured in LB medium at 37 °C. When the OD600 reached 0.6-0.8, isopropyl β-D-1-thiogalactopyranoside (IPTG, 0.2 mM) was added to cells to induce protein expression at 18 °C for 18-20 h. Then, the cells were harvested by centrifugation at 6000 rpm for 15 min at 4 °C. The cell pellets were resuspended in a lysis buffer (50 mM Tris-HCl pH 8.0, 500 mM NaCl, 10 mM imidazole pH 8.0) and disrupted using a high-pressure homogenizer (JNBIO, China). The cell debris was removed by centrifugation at 17,000 rpm for 60 min at 4 °C. Next, the supernatant was purified using a His TrapTM HP column (GE Healthcare) and was eluted in a buffer (50 mM Tris-HCl, 500 mM NaCl, 500 mM imidazole pH 8.0). The His-sumo tag was removed by the ULP1 enzyme cleavage, followed by additional Ni-NTA affinity chromatography as previously described. The protein was further purified by gel filtration using a Superdex200 16/600 column (GE Healthcare) in a buffer (20 mM Tris-HCl pH 8.0, 100 mM NaCl).
Crystallization and data collection
The E12 and E1CTD-E12 complexes were crystallized using the sitting-drop vapor-diffusion method by mixing 0.2 μL protein and 0.2 μL reservoir solution at 18 °C. E12 crystals were grown in a reservoir solution containing 2.0 M ammonium sulfate, 0.1 M CAPS/sodium hydroxide pH 10.5, 0.2 M lithium sulfate with a protein concentration of 15 mg/mL. For the E1CTD-E12 complex, crystals were grown in a reservoir solution containing 1600 mM sodium phosphate monobasic/400 mM potassium phosphate dibasic, 100 mM sodium phosphate dibasic/citric acid pH 4.2 with a protein concentration of 20 mg/mL. For the E1CTD-E12 (H122D)-SAM complex, a mixture of E1CTD-E12 (H122D) and SAM (Aladdin) at a molar ratio of 1:5 was prepared and incubated on ice for 1 h before performance of crystallization experiments. The E1CTD-E12 (H122D)-SAM complex crystals were grown in a reservoir solution containing 0.1 M sodium acetate trihydrate pH 4.6, 2.0 M sodium formate with a protein concentration of 20 mg/mL. All crystals were briefly soaked in a cryoprotectant solution consisting of 25% (v/v) glycerol dissolved in their corresponding mother liquors before being flash-cooled directly in a liquid-nitrogen stream at 100 K. The X-ray diffraction data were collected at the beamlines of the Shanghai Synchrotron Radiation Facility. The process of data integration, scaling, and merging was performed using the HKL3000 package and the XDS programme [Citation24].
Structure determination and refinement
The crystal structure of MPXV E12 was determined by molecular replacement using AlphaFold2-predicted E12 structure (https://alphafold.ebi.ac.uk) as a search model. E1CTD-E12 and E1CTD-E12 (H122D)-SAM complex structures were determined by molecular replacement using the crystal structure of the VACV mRNA MTase structure (PDB: 2VDW) as the search model. Cycles of refinement and model building were carried out by using Phenix and COOT programmes [Citation25]. All of the structures were displayed and analyzed using the PyMOL programme. The collected data and refinement statistics are summarized in .
Dynamic light scattering (DLS) measurement
The DLS data were collected using the DYNAMICS software from DynaPro NanoStar (Wyatt Technology), operating at a light source wavelength of 658 nm and a fixed scattering angle of 90°. E12 and E1CTD-E12 were diluted to 1 mg/mL with a buffer containing 20 mM Tris-HCl (pH 8.0) and 100 mM NaCl at 25 °C.
Small angels X-ray scattering (SAXS)
The SAXS data of E12 and E1CTD-E12 were collected at beamline BL19U2 of the Shanghai Synchrotron Radiation Facility, with a radiation wavelength of 1.03 Å. The protein samples were prepared at 0.1-2 mg/mL concentrations in the buffer (20 mM Tris-HCl pH 8.0, 150 mM NaCl). The samples were measured in triplicate, and the sample measurements were adjusted by subtracting the scattering from the buffer alone. Data were analyzed using the software package BioATSAS (https://www.embl-hamburg.de/biosaxs/). The scattering images were averaged and subtracted from the buffer scattering images. Then, using the indirect Fourier transform method, the Rg was estimated. The distribution function p(r) was calculated from the parameter Dmax. The SAXS envelopes of the E12 and E1CTD-E12 complex were built by GASBOR, as previously reported [Citation25].
Isothermal titration calorimetry (ITC) assay
The ITC measurement was performed using MICROCAL PEAQ-ITC (Malvern) at 25 °C. 20 μM E1CTD was titrated with 100 μM E12. 20 μM E1CTD, 20 μM E1CTD-E12, and 20 μM Δ(545-562) E1CTD-E12 were titrated with 200 μM SAM, respectively. The fitting curve with a single-binding-site model was performed by the ITC data analysis module provided by the manufacturer.
Surface plasmon resonance (SPR) assay
The SPR binding experiments were carried out using a Biacore 8 K (Cytiva) at 25 °C in the single-cycle mode. All proteins for SPR assays were exchanged to PBST buffer (10 mM Na2HPO4, 2 mM KH2PO4, pH 7.4; 137 mM NaCl; 2.7 mM KCl, and 0.05% Tween-20). The E1CTD was immobilized on the CM5 chip and gradient concentrations of E12 (from 781.25–25000 nM) was then used to flow over the chip surface at 30 μl min−1 and the real-time response was recorded. After each cycle, the sensor surface was regenerated using 10 mM glycine-HCl (pH 2.5). KD values for SPR assays were calculated using a 1:1 binding fit model with the software BIAevaluation version 4.0.8 (Cytiva). The SPR binding assays were conducted three times, and the proteins from different batches were used in the replication experiments.
Author contribution statement
J.L. and J.D. conceived and designed this study. A.C., N.F., Z.Z., Y.W., Y.S., Y.Z., L.Z., and G. Z. performed research and analyzed data; J.L. and A.C. wrote the manuscript. All authors discussed the results and commented on the manuscript.
Supplemental Material
Download MS Word (1.2 MB)Acknowledgments
We thank the staff of the Shanghai Synchrotron Radiation Facility beamlines BL02U1, BL10U2, BL18U1, BL19U1, and BL19U2 for help with X-ray data and SAXS data collection.
Data availability
The coordinates and structural factors generated in this study have been deposited in the Protein Data Bank with accession codes 8Y2Y (E12) [https://www.rcsb.org/structure/8Y2Y], 8Y2Z (E1CTD-E12 complex) [https://www.rcsb.org/structure/8Y2Z], and 8ZE4 (E1CTD-E12 (H122D)-SAM complex) [https://www.rcsb.org/structure/8ZE4]. The SAXS data and model have been deposited in the SASBDB database with accession codes SASDUL2 for E12 and SASDUM2 for the E1CTD-E12 complex. Source data are provided in this paper.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Lum F-M, Torres-Ruesta A, Tay MZ, et al. Monkeypox: disease epidemiology, host immunity and clinical interventions. Nat Rev Immunol. 2022;22(10):597–613. doi:10.1038/s41577-022-00775-4
- Luo YH, Zhang T, Cao JL, et al. Monkeypox: an outbreak of a rare viral disease. J Microbiol Immunol Infect. 2023.
- Khan SA, Parajuli SB, Rauniyar VK. Neurological manifestations of an emerging zoonosis-Human monkeypox virus: a systematic review. Medicine (Baltimore). 2023;102(35):e34664.
- Rampogu S, Kim Y, Kim S-W, et al. An overview on monkeypox virus: pathogenesis, transmission, host interaction and therapeutics. Front Cell Infect Microbiol. 2023;13:1076251, doi:10.3389/fcimb.2023.1076251
- Shatkin AJ. Capping of eucaryotic mRNAs. Cell. 1976;9(4 PT 2):645–653.
- Furuichi Y, Shatkin AJ. Viral and cellular mRNA capping: past and prospects. Adv Virus Res. 2000;55:135–184.
- Decroly E, Ferron F, Lescar J, et al. Conventional and unconventional mechanisms for capping viral mRNA. Nat Rev Microbiol. 2011;10(1):51–65.
- Shchelkunov SN, Totmenin AV, Safronov PF, et al. Analysis of the monkeypox virus genome. Virology. 2002;297(2):172–194. doi:10.1006/viro.2002.1446
- Kyrieleis OJP, Chang J, de la Peña M, et al. Crystal structure of vaccinia virus mRNA capping enzyme provides insights into the mechanism and evolution of the capping apparatus. Structure. 2014;22(3):452–465. doi:10.1016/j.str.2013.12.014
- Carpenter MS, DeLange AM. A temperature-sensitive lesion in the small subunit of the vaccinia virus-encoded mRNA capping enzyme causes a defect in viral telomere resolution. J Virol. 1991;65(8):4042–4050. doi:10.1128/jvi.65.8.4042-4050.1991
- De la Pena M, Kyrieleis OJ, Cusack S. Structural insights into the mechanism and evolution of the vaccinia virus mRNA cap N7 methyl-transferase. EMBO J. 2007;26(23):4913–4925. doi:10.1038/sj.emboj.7601912
- Martin JL, McMillan FM. SAM (dependent) I AM: the S-adenosylmethionine-dependent methyltransferase fold. Curr Opin Struct Biol. 2002;12(6):783–793. doi:10.1016/S0959-440X(02)00391-3
- Fabrega C, Hausmann S, Shen V, et al. Structure and mechanism of mRNA cap (guanine-N7) methyltransferase. Mol Cell. 2004;13(1):77–89. doi:10.1016/S1097-2765(03)00522-7
- Benarroch D, Smith P, Shuman S. Characterization of a trifunctional mimivirus mRNA capping enzyme and crystal structure of the RNA triphosphatase domain. Structure. 2008;16(4):501–512. doi:10.1016/j.str.2008.01.009
- Du X, Gao ZQ, Geng Z, et al. Structure and biochemical characteristic of the methyltransferase (MTase) domain of RNA capping enzyme from African swine fever virus. J Virol. 2021;95(5.
- Higman MA, Christen LA, Niles EG. The mRNA (guanine-7-)methyltransferase domain of the vaccinia virus mRNA capping enzyme. Expression in Escherichia coli and structural and kinetic comparison to the intact capping enzyme. J Biol Chem. 1994;269(21):14974–14981. doi:10.1016/S0021-9258(17)36562-6
- Ramanathan A, Robb GB, Chan S-H. mRNA capping: biological functions and applications. Nucleic Acids Res. 2016;44(16):7511–7526. doi:10.1093/nar/gkw551
- Ghosh A, Lima CD. Enzymology of RNA cap synthesis. Wiley Interdiscip Rev RNA. 2010;1(1):152–172. doi:10.1002/wrna.19
- Ho CK, Gong C, Shuman S. RNA triphosphatase component of the mRNA capping apparatus of Paramecium bursaria Chlorella virus 1. J Virol. 2001;75(4):1744–1750. doi:10.1128/JVI.75.4.1744-1750.2001
- Shuman S, Hurwitz J. Mechanism of mRNA capping by vaccinia virus guanylyltransferase: characterization of an enzyme–guanylate intermediate. Proc Natl Acad Sci U S A. 1981;78(1):187–191. doi:10.1073/pnas.78.1.187
- Imprachim N, Yosaatmadja Y, Newman JA. Crystal structures and fragment screening of SARS-CoV-2 NSP14 reveal details of exoribonuclease activation and mRNA capping and provide starting points for antiviral drug development. Nucleic Acids Res. 2023;51(1):475–487. doi:10.1093/nar/gkac1207
- Dong H, Ray D, Ren S, et al. Distinct RNA elements confer specificity to flavivirus RNA cap methylation events. J Virol. 2007;81(9):4412–4421. doi:10.1128/JVI.02455-06
- Ahola T, Kaariainen L. Reaction in alphavirus mRNA capping: formation of a covalent complex of nonstructural protein nsP1 with 7-methyl-GMP. Proc Natl Acad Sci U S A. 1995;92(2):507–511. doi:10.1073/pnas.92.2.507
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi:10.1016/S0076-6879(97)76066-X
- Zhan B, Gao Y, Gao W, et al. Structural insights of the elongation factor EF-Tu complexes in protein translation of Mycobacterium tuberculosis. Commun Biol. 2022;5(1):1052, doi:10.1038/s42003-022-04019-y