
Abstract
Enterovirus A71 (EV-A71) causes Hand, Foot, and Mouth Disease and has been clinically associated with neurological complications. However, there is a lack of relevant models to elucidate the neuropathology of EV-A71 and its mechanism, as the current models mainly utilize animal models or immortalized cell lines. In this study, we established a human motor neuron model for EV-A71 infection. Single cell transcriptomics of a mixed neuronal population reveal higher viral RNA load in motor neurons, suggesting higher infectivity and replication of EV-A71 in motor neurons. The elevated RNA load in motor neurons correlates with the downregulation of ferritin-encoding genes. Subsequent analysis confirms that neurons infected with EV-A71 undergo ferroptosis, as evidenced by increased levels of labile Fe2+ and peroxidated lipids. Notably, the Fe2+ chelator Deferoxamine improves mitochondrial function and promotes survival of motor neurons by 40% after EV-A71 infection. These findings deepen understanding of the molecular pathogenesis of EV-A71 infection, providing insights which suggest that improving mitochondrial respiration and inhibition of ferroptosis can mitigate the impact of EV-A71 infection in the central nervous system.
Disclaimer
As a service to authors and researchers we are providing this version of an accepted manuscript (AM). Copyediting, typesetting, and review of the resulting proofs will be undertaken on this manuscript before final publication of the Version of Record (VoR). During production and pre-press, errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal relate to these versions also.Introduction
Enterovirus A71 (EV-A71) is an RNA virus belonging to the Picornaviridae family, and is known for causing hand, foot, and mouth disease (HFMD), primarily affecting young children. EV-A71 is characterized by its ability to spread rapidly, leading to outbreaks in various regions globally. While HFMD symptoms are generally mild, EV-A71 can, in some cases, result in severe life-threatening neurological complications, including meningitis and encephalitis [1,2]. Even though EV-A71 is hypothesized to replicate in skeletal muscles, subsequently infecting motor neurons at the neuromuscular junction and eventually reaching the central nervous system via retrograde axonal transport [3-5], the molecular mechanisms involved in EV-A71 neuropathogenesis remain unclear. This is compounded by the lack of appropriate humanized disease models which impeded the study of EV-A71’s neuropathogenesis and pathology.
Current studies on EV-A71 pathogenesis rely heavily on animal models and immortalized human cell lines such as SH-SY5Y. While murine models often offer valuable insights into the biology of infectious diseases, species-specific differences remain and they are not fully representative of human cells [6]. For instance, while human scavenger receptor class B member 2 (SCARB2) serves as a receptor for EV-A71 entry, the mouse Scarb2 does not fulfil this function [7,8]. Conversely, in vitro models enable the study of human neural cell host responses. Although human neuroblastoma cell lines and neural progenitor cells (NPCs) have been utilized [9], they significantly differ from actual human neurons, both in terms of gene expression, cellular function and response to stressors.
To address this gap in the availability of humanized in vitro models of EV-A71 neuropathogenesis, we explored the use of neurons derived from human pluripotent stem cells as a model for studying EV-A71 biology. Human pluripotent stem cells, including embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs), offer significant advantages in modelling human diseases by allowing the unlimited generation of human cell types, such as neurons, which are challenging to obtain directly from patients. Using this strategy, we showed that human ESC-derived motor neurons are susceptible to EV-A71 infection, which gave us an opportunity to investigate motor neuron-intrinsic host cell responses. Within a mixed population of neural cell types, single cell transcriptomics revealed that viral RNA load was highest within the motor neuron population. EV-A71-infected motor neurons subsequently displayed mitochondrial dysfunctions and increase in intracellular free iron, resulting in ferroptosis.
This yielded important insights into the biology of EV-A71 neuropathogenesis that were previously unknown. By sequestering free iron, we demonstrated that mitochondrial defects and motor neuron death were rescued, suggesting that inhibition of ferroptosis could be an important therapeutic strategy. Together, our findings demonstrate the interaction between EV-A71 infection and ferroptosis, and provide novel insights into neurodegeneration induced by EV-A71.
Materials and Methods
Virus strains and propagation
The EV-A71 virus was propagated in human rhabdomyosarcoma (RD) cells. RD cells were cultured in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS). The EV-A71 S41 strain (5865/SIN00009, Accession No. AF316321, sub-genogroup B4) was used in most experiments in this study unless otherwise specified. S41 was derived from a clinical isolate that caused encephalitis, interstitial pneumonitis, and pulmonary edema [10], In additional experiments, the MS strain (EV71/7423/MS/87, Accession No. U22522, sub-genogroup B2) and C2 strain (NUH0075/SIN/08, Accession No. FJ172159, sub-genogroup C2) were used alongside the S41 strain. MS isolate was obtained from a patient who displayed acute flaccid paralysis [11], while C2 was obtained from an uncomplicated HFMD case [12].
Plaque assay
RD cells were plated at a density of 500,000 cells per well in a 6-well plate. The following day, supernatants from infected cells were serially diluted in DMEM and added to RD cells (0.5 mL per well) for 1 h at 37°C. Subsequently, low-melting-point agarose, prepared in DMEM supplemented with 10% FBS, was added to each well and allowed to gel at room temperature before transfer to the incubator for 3 days. Thereafter, the cells were fixed with 4% formaldehyde and stained with 0.1% crystal violet solution. After washing, plaque numbers were counted. Viral titers were expressed as plaque-forming units per milliliter (PFU/ml).
Embryonic stem cell maintenance
H7 embryonic stem cells (ESCs) were maintained on Matrigel-coating with StemMACS™ iPS-Brew XF (Miltenyi Biotec) in a humidified incubator at 37°C with 5% CO2. The cells were passaged every 5-7 days upon reaching 70% confluence using ReLeSR (Stem Cell Technologies).
Spinal motor neuron differentiation
H7 ESCs were differentiated toward motor neurons using a well-established protocol as previously published [13-15]. Neural induction of ESCs began with N2B27 medium supplemented with CHIR99021 (4.25 µM) and LDN193189 (0.5 µM). The N2B27 medium comprised DMEM/F12: Neuro Medium (50:50), 1× Neurobrew supplement, 1× N2 supplement, 1× Non-Essential Amino Acids, and 1× L-glutaMAX. On day 3, retinoic acid (1 µM) was introduced to the neural induction medium, and the cells were maintained until day 10. On day 10, the progenitor cells were either frozen for future use or further differentiated with 1 µM retinoic acid and 2 µM sonic hedgehog agonist, purmorphamine. From day 17 to day 28, the medium was changed to maturation medium (N2B27 supplemented with 200 µM ascorbic acid, 1% sodium pyruvate, 10ng/ml BDNF, and 10ng/ml GDNF).
Infection
On day 27, cells were reseeded to the appropriate density for infection. On day 28, the cells were infected with EV-A71 (S41 strain) at different MOIs. During infection, the medium was removed, and EV-A71 prepared at different MOIs was added directly to the cells, allowing for a 1-hour adsorption period. After 1 hour, the virus was removed by washes, and fresh N2B27 medium was added to the cells. The cells and culture supernatants were then harvested at various hours post-infection (hpi).
Immunostaining Assay
At each MOI, the supernatant was collected for plaque assays or further analysis. The cells were fixed in 4% paraformaldehyde for 15 minutes at room temperature. For immunostaining, the cells were permeabilized with 0.1% Triton-X for 15 minutes at room temperature. Subsequently, a blocking buffer (5% FBS + 1% BSA in PBS) was added and incubated for 1 hour. The primary antibody (Table S1) was diluted accordingly in the blocking buffer and incubated overnight at 4 degrees Celsius. On the next day, cells were washed twice before adding secondary antibodies (Table S1) and DAPI for 1.5 hours. Afterward, the cells were washed with PBS and imaged using an Opera Phenix High-Content Screening System. All image analyses were performed using Columbus software based on a predefined pipeline.
Single cell RNA-Seq
EV-A71-infected (MOI 5) and mock-infected cells were harvested at 6 hpi for scRNA-Seq. Cells were detached by accutase treatment for 10 minutes at 37°C and single-cell suspensions were resuspended in PBS+0.02% BSA. Single-cell RNA sequencing libraries were prepared using 10x genomics Chromium Single cell 3 prime workflow and sequenced in Novogene 6000. For 10X analyses, alignment and UMI counting were performed by using the Cellranger program with default parameters and human hg38 reference genome plus S41 complete genome (AF316321). Feature-barcode count matrix obtained from Cellranger was used for the downstream analyses using Seurat in R. For the quality control, cells whose number of expressed genes <1000, and percentage of mitochondrial genes > 10 were filtered out. Genes expressed in less than three cells were removed. UMAP was used to do the visualization with 20 dimensions after dimension reduction using PCA. Differentially expressed genes in each cluster or cell type were identified using FindAllMarkers. Genes that were differentially expressed between Mock and S41 were identified by FindMarkers using Wilcoxon's rank sum tests with a minimum upregulation of 0.25 log-fold and a p-value of 0.05. DoHeatmap was used to visualize the top markers for each lineage. The S41 sample cells were classified into EV-A71-virus high and low cells based on the cutoff (3) 75% quantiles of S41 expression. The differential genes between the virus -high and virus-low groups for each cell type were identified using Wilcoxon's rank sum tests with a minimum upregulation of 0.1 log-fold and a p-value cutoff of 0.01. Gene ontology analysis was performed using Metascape (http://metascape.org) and GO terms were visualized in R using ggplot2. The scRNA-seq datasets have been deposited and are available in the NCBI Gene Expression Omnibus (GEO) database under the accession number GSE261751.
Organoid Assay
Spinal cord organoids were generated from H7 ESCs following previously published protocols [14,16]. On day 30 of differentiation, the organoids were infected with 104 PFU/mL of EV-A71 (S41 strain). After 24 hours, the virus was removed by aspirating the culture media, and the organoids were washed. Fresh media was added, and the organoids were further incubated for another 2 days. Subsequently, the organoids were fixed in 4% paraformaldehyde, cryo-sectioned, and immunostained for the respective markers. Images were captured using a Nikon Ti-E fluorescence microscope, and image analysis was performed using ImageJ.
Measurements of mitochondrial membrane potential (MMP)
The JC-1 assay (Invitrogen) was performed to evaluate mitochondrial membrane potential following the manufacturer's protocol. Briefly, the cell medium was removed, and the cells were washed once with PBS. Subsequently, the PBS was replaced with JC-1 dye diluted in N2B27 medium (2 µM working concentration), and the cells were incubated at 37°C for 15 minutes. Afterward, they were washed twice with N2B27 medium and analyzed by a plate reader using wavelengths at Ex/Em = 514 nm/530 nm for monomer and 514 nm/590 nm for J-aggregate. For positive controls, Carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP) was added at a working concentration of 100 µM after JC-1 labeling. For drug treatment, elamipretide (SS-31, Selleckchem) was reconstituted in distilled water and added to the medium post-infection at concentrations ranging from 100 nM to 10 µM.
FerroOrange Assay
To assess intracellular free iron (Fe2+), the FerroOrange assay was performed following the manufacturer's protocol (Dojindo). Briefly, cells were washed three times with Hank's Balanced Salt Solution (HBSS). Subsequently, FerroOrange solution was diluted in HBSS to prepare a working solution of 200 nM FerroOrange, which was then added to the cells. The cells were incubated for 30 min at 37°C before being imaged with wavelengths at Ex/Em = 561 nm/599 nm. Hoechst-33342 was used as a counterstain to visualize the nuclei.
Lipid peroxidation assay
To assess lipid peroxidation, BODIPY™ 581/591 C11 (Invitrogen) was used at the fluorescent probe to detect oxidized lipids. Cells were incubated with the BODIPY 581/591 C11 dye diluted in HBSS at a concentration of 1 µM, for 30 min, before being washed twice with HBSS. The cells were then imaged at 63× using confocal microscopy (Opera Phenix) with wavelengths at Ex/Em = 488 nm/522 nm and Ex/Em = 561 nm/599 nm.
Ferroptosis inhibitors treatment
Ferrastatin-1 and Deferoxamine (DFO) (both from Selleckchem) were reconstituted in DMSO and diluted in N2B27 medium to specified concentrations. Cells were treated with drugs for at least 1 hour before the addition of the virus. After a 1-hour adsorption period, the virus was removed and fresh medium with drugs was added. At respective time points post-infection, the cells were then analyzed for infection rate, cell viability (by DAPI count), and plaque assays.
Gene knockdown and qPCR
For gene knockdown experiments, hESC-derived motor neurons were seeded overnight at a density of 750,000 cells (12-well plates) or 60,000 cells (96-well plates). On the next day, the cells were transfected with the respective siRNAs using Lipofectamine RNAiMAX at a concentration of 40 nM. At 48 hours post-transfection, RNA was extracted using TRIzol, followed by reverse transcription and qPCR (primers are listed in Table S2). Similarly, the cells were infected with EV71 S41 at 48 hours post-transfection and harvested at respective time points (6 hpi, 24 hpi, or 48 hpi) for the corresponding analyses. The siRNAs used were: SCARB2 (Dharmacon, #012087), FTL (SCBT, sc-40577), FTH1 (SCBT, sc-40575).
Statistical analysis
All results were analyzed using GraphPad Prism software. Normality tests were conducted, and for normally distributed data, t-tests were employed for comparisons between two groups, while one-way ANOVA was used for comparisons among more than two groups. Non-normally distributed data were analyzed using the Mann-Whitney U test for comparisons between two groups and the Kruskal-Wallis (KW) test for comparisons among more than two groups. Each experiment was repeated at least thrice, and results were normalized to the control for each experiment when necessary. Values are presented as means ± SD. A p-value below 0.05 was considered statistically significant. Details for each test and condition are further indicated in the legend of each figure.
Results
Human ESC-derived neural cultures are susceptible to EV-A71 infection
To create a cell culture-based model with human relevance, we differentiated human pluripotent stem cells (in this case hESCs) into a mixed population of spinal neurons and progenitor cells, using a well-established protocol [13-15]. At the end of the 28-day protocol, ISL1+SMI32+ motor neurons (MNs) were detected, along with CHX10+ and Calbindin+ interneurons and small numbers of SOX1+ neural progenitor cells (NPCs) (Figure S1). To determine if the hESC-derived neuronal cultures were susceptible to EV-A71 infection, day 28 cultures were infected at MOI 1 and 5, and cultures were subsequently analyzed at 6, 24, 48, and 72 hours post infection (hpi) (A). In general, we found that hESC-derived neurons were susceptible to EV-A71 infection, and that infectivity increases with higher MOI ((B-D), up to 49.1 ± 14.1 % at 72hpi with MOI 5. The infectivity of EV-A71 in the human neurons is lower than SH-SY5Y cells (Figure S2) but similar to NSC-34 cells [17]. Cytopathic effect (CPE) with rounded cells lifted from the plate was evident at 24 hours when infected at MOI 5, while the same was observed at 48 hours when infected at MOI 1. (Figure S3). The high number of dead cells at 72 hpi could likely confound the analysis of the infected ISL1+ MNs with MOI 5 ((D). Additionally, the virus within the cells was actively replicating, as verified by the detection of non-structural protein EV-A71 3D, and dsRNA, an intermediate of viral genome replication ((E). Furthermore, increased viral titers over time measured in the culture supernatants further supported that EV-A71 replicates productively in this culture system ((F).
Figure 1. Human ESC-derived motor neurons are susceptible to EV-A71 infection. A. hESC were differentiated for 28 days into motor neurons and then were infected with EV-A71 S41. B. Immunodetection of EV-A71 VP2 viral capsid protein. White arrows indicate cells where VP2 and ISL1 staining are colocalized. C. Percentage of VP2+ cells and D. VP2+ ISL1+ MNs, detected at multiplicities of infection (MOI) 1 and 5, n=6. Quantification at 72 hpi might be inaccurate due to the presence of floating dead cells. E. Immunostaining for non-structural viral protein (EV-A71 3D) and dsRNA. F. Virus titers in the supernatant was determined by plaque assay using RD cells, n=3.
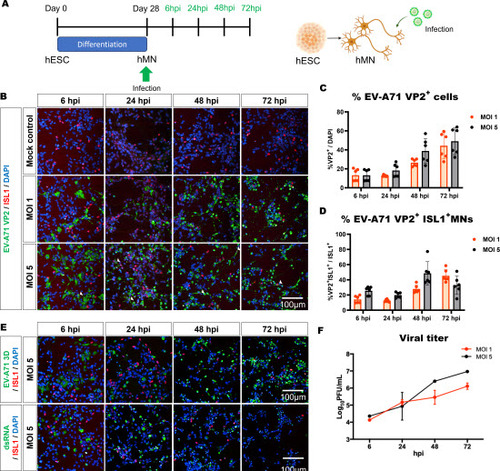
We also infected the hESC-derived neuronal cultures with two other EV-A71 clinical isolates, namely MS and C2, which are myotropic strains associated with milder symptoms (MS: Acute flaccid paralysis; C2: Uncomplicated), compared to the fatal neurovirulent S41 strain (Encephalitis) [17,18]. All three clinical isolates/viral strains infected hESC-derived neurons at a similar rate and showed similar viral kinetics even though the patients exhibited different clinical symptoms (Figure S4) and the strains showed different infectivity in NSC-34 cells [17]. To validate that hESC-derived neuronal cultures are a valid model of EV-A71 infection, we targeted SCARB2, a known human-specific host receptor, using siRNA and show that SCARB2 knockdown leads to lower infection rates at 6 hpi (Figure S5A-C). Furthermore, previously, we have shown that Rocaglamide A (RocA) is a small molecule inhibitor of Prohibitin, a host target of EV-A71 [18]. We further demonstrate here that RocA treatment reduced EV-A71 infection in hESC-derived neurons (Figure S5D&E). Taken together, our data validate human pluripotent stem cells as a suitable model for studying the biology of EV-A71 infection in human neurons.
Single cell transcriptomics reveal highest EV-A71 RNA load within MNs
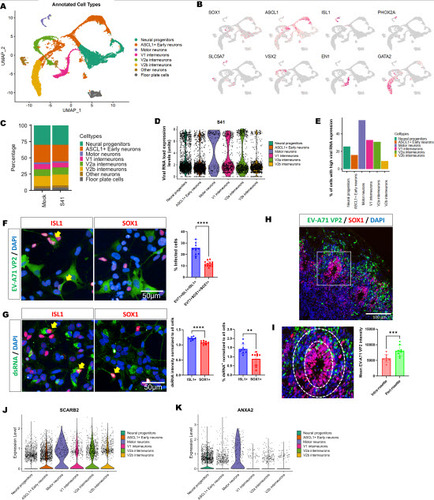
To explore the host gene expression dynamics during EV-A71 infection, we infected the hESC-derived spinal neuronal cultures with EV-A71 (S41 strain) at MOI 5, and harvested these, as well as the mock-infected controls, at 6 hpi for single cell RNA-sequencing (scRNA-seq) using the 10x Genomics platform. First, unsupervised clustering analysis of the scRNA-seq results unveiled the presence of distinct cell populations, namely motor neurons, neural progenitors, V1 interneurons, V2a interneurons, V2b interneurons, early neurons, other neurons and floor plate cells (A). These cell populations exhibited expression profiles consistent with their respective molecular markers (B, Figure S6). Although EV-A71 infection did not exert a global influence on the composition and proportion of different cell types at 6 hpi (C), we noticed variations in viral RNA load within each cell population from the scRNA-seq results (D). Notably, motor neurons exhibited higher viral RNA load compared to other neural cell types, implying that motor neurons are more susceptible to EV-A71 S41 infection than other cell types (D-E). Indeed, immunostaining of neural cultures infected with EV-A71 revealed that a higher percentage of ISL1+ motor neurons were positive for EV-A71 VP2 (25.8 ± 7.3 %) compared to SOX1+ neural progenitor cells (11.8 ± 2.4 %). (F). Similarly, the signal intensity for dsRNA was also significantly higher in motor neurons as compared to neural progenitors (G), suggesting higher viral replication within motor neurons. We verified this using a 3D spinal cord organoid model we developed previously [14,16] and observed that while EV-A71 infected the neural progenitors and neurons within the organoids (Figure S7), the EV-A71 VP2 signals within the SOX1+ neural rosettes were lower (H-I), corroborating our findings from the 2D culture. We also found that motor neurons had higher expressions of EV-A71 host factors, SCARB2 and ANXA2 in both uninfected (J-K) and infected cultures (Figure S8). Collectively, we determined that motor neurons are more permissive to EV-A71 infection and replication with the use of single cell transcriptomics.
Figure 2. scRNA-Seq analysis to study host gene expression during EV-A71 infection reveal highest viral RNA in MNs within a heterogeneous culture. A. Annotated Seurat plot showing the presence of motor neurons, spinal interneurons, early neurons and neural progenitor cells within the human ESC-derived spinal neural culture. B. Distribution of expressed markers in each population. C. EV-A71 infection has no global effect on composition of the neural culture at 6 hpi. D. Expression level of EV-A71 S41 RNA in each cell population. E. Percentage of “virus-high” cells in each population. “Virus-high” cells are defined by cells having viral RNA that is higher than the mean expression level. F. Immunostaining of neural cultures infected with EV-A71 using antibodies against EV-A71 VP2, revealing higher percentage of EV-A71 infected ISL1+ MNs than SOX1+ NPCs. n=12, t-test, ****p<0.0001. Yellow arrows indicate double-positive cells. G. Higher double-stranded RNA (dsRNA) signals and percentage of dsRNA+ cells within ISL1+ MNs as compared to SOX1+ NPCs. n=12, t-test, ****p<0.0001. Yellow arrows indicate double-positive cells. H. Immunostaining of infected spinal cord organoids. I. Magnified images of a rosette of the infected spinal cord organoids. Quantification of the EV-A71 VP2 intensity within the SOX1+ neural rosette and outside the rosette. n=8, paired t-test, ***p<0.001. J. Expression level of SCARB2 RNA in each cell population (mock). K. Expression level of ANXA2 RNA in each cell population (mock).
Gene expression changes within EV-A71-infected MNs suggest mitochondrial dysfunction
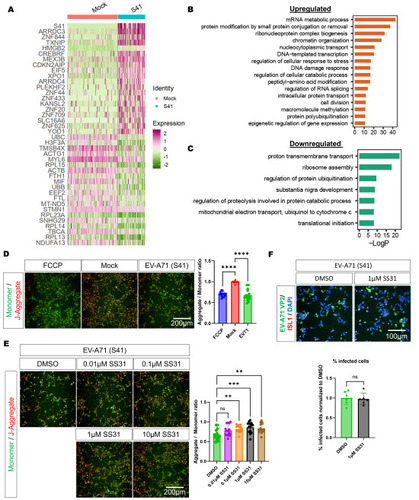
To uncover the gene expression changes between infected and mock-infected motor neurons, we conducted a differential gene expression analysis, which identified top upregulated genes such as ARRDC3, ZNF844, and TXNIP (A, Supplementary data file 1). Many of these upregulated genes were also common in the other neural cell populations (Figure S9). Next, we conducted Gene Ontology analyses on EV-A71-infected vs mock-infected motor neurons to elucidate enrichments of specific gene sets following infection and found that upregulated gene sets were mainly associated with the transcriptional regulation, protein modifications, and RNA splicing (B), which are in concordance with a previously published work [19]. Using the same analysis parameters, we found that the top downregulated gene sets are related to mitochondrial function (C), and the downregulation of genes involved in proton transmembrane transport and mitochondrial electron transport suggests that EV-A71-infected motor neurons have dysfunctional mitochondria. These findings were consistent across various neural cell types (Figure S10) as well as when comparing cells with high viral RNA load (“virus-high” cells) and cells with low viral RNA load (“virus-low” cells) (Figure S11). To validate that EV-A71-infected motor neurons indeed have reduced capacity for mitochondrial respiration, we performed a JC-1 assay and observed reduction of MMP in EV-A71-infected cultures, at 24 hpi (D). Next, to investigate if the reduction in MMP is reversible, we treated the EV-A71-infected neurons with elamipretide (SS31), which is known to stabilize cardiolipin in the inner mitochondrial membrane [20], and found a partial restoration of the MMP (E). However, SS31 treatment did not alter the infection dynamics of EV-A71 (F).
Figure 3. DEGs of the infected cells suggest mitochondrial dysfunction. A. Top up- and down-regulated genes for motor neurons after EV-A71 infection as compared to mock-infected cells. B. Gene Ontology enrichment analysis of the significantly upregulated genes after EV-A71 infection in MNs. C. Gene Ontology enrichment analysis of the significantly downregulated genes after EV-A71 infection in MNs. D. JC-1 assay was conducted to validate occurrence of mitochondrial dysfunction after infection. n=15, One-way ANOVA, ****p<0.0001. E. Effects of treatment with elamipretide (SS31) on mitochondrial function and EV-A71 infection. n=15, One-way ANOVA, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. F. SS31 treatment did not lead to significant changes of infection rate. n=6, Student’s t-test.
Motor neurons infected with EV-A71 undergo ferroptosis
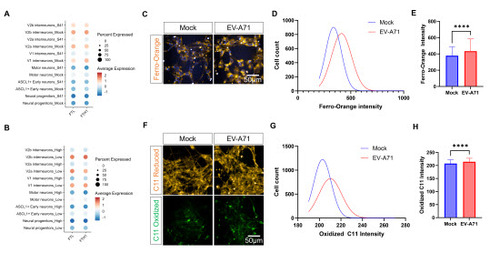
Since we found that SS31 treatment was not sufficient to rescue the cells from EV-A71-induced cell death, we further investigated other downregulated genes that could be related to mitochondrial dysfunction. Interestingly, we found that several genes, including FTL and FTH1, consistently appeared among the top downregulated genes after infection in both motor neurons and ventral interneurons, as compared to their respective mock infection controls (Figure S12A, 4A, S12B). Expanding on this finding, we also observed downregulation of FTL and FTH1 in the “virus-high” motor neurons versus those the “virus-low” motor neurons (B, S12C). FTL and FTH1 encode the light and heavy chains of ferritin respectively. Ferritin is an important iron storage protein, and downregulation of ferritin has been linked to elevated intracellular labile iron pool [21-23]. We postulate that the decrease in FTL and FTH1 after EV-A71 infection would then result in ferroptosis, which is further supported by changes in the expression of ferroptosis-associated genes, such as GPX4 and TFRC, both after infection and in the “virus-high” cells (Figure S12B-C). Notably, TFRC encodes transferrin receptor protein 1, and its upregulation has been shown to promote ferroptosis in Coxsackievirus B3 infection by importing extracellular iron into the cells [24,25]. To confirm that EV-A71 infection results in ferroptosis, we first used the Ferro-Orange assay to determine the levels of intracellular free iron after EV-A71 infection. Ferro-Orange is a fluorescent probe that specifically detects labile Fe2+ ions, and we observed an increase in Ferro-Orange signals in hESC-derived neurons infected with EV-A71 (433.8 ± 149.5 A.U. versus 378.4 ± 106.7 A.U. in mock; p-value < 0.0001) (C-E). Next, we assessed levels of peroxidated lipids using BODIPY C11 staining and saw a marked increase in C11 oxidation following infection, indicating increased lipid peroxidation in response to the viral infection (F-H). The increase in Ferro-Orange intensity and C11 oxidation is similar to the effect of treatment with the known ferroptosis inducer, Erastin, on the hESC-derived neurons (Figure S13). At the same time, we also investigated the effects of FTH1 and FTL downregulation on motor neurons. Using specific siRNAs against FTH1 and FTL, we demonstrated simultaneous knockdown of both genes. This resulted in elevated Ferro-Orange signals similar to what was observed for EV-A71 infection. Following the siRNA treatment, we observed reduced motor neuron survival, suggesting that loss of ferritin triggers iron-dependent motor neuron death (Figure S14). These results confirmed that EV-A71 infection leads to ferroptosis, which is characterized by an increased labile iron pool and increased lipid peroxidation and is likely driven by loss of ferritin.
Figure 4. Ferroptosis in human motor neurons after EV-A71 infection. A. Expression levels of FTL and FTH1 after infection in each population. B. Expression levels of FTL and FTH1 after infection in each population with high viral RNA load (High) and low viral RNA load (Low). C. Free irons within the cells were measured by Ferro-Orange. D. Number of cells that were positive for Ferro-Orange in the EV-A71-infected and mock-infected cultures. E. Ferro-Orange intensity in each cell at 6 h after EV-A71 or mock infection. n>1900 cells from 3 cultures, Mann-Whitney Test, ****p<0.0001. F. Lipid peroxidation measured by BODIPY C11 581/591. G. Number of cells displaying oxidized C11 in EV-A71-infected and mock cultures. H. Mean oxidized C11 intensity in each cell at 24 h after EV-A71 or mock infection. n>2300 cells from 3 cultures, Mann-Whitney Test, ****p<0.0001.
Deferoxamine (DFO) rescues mitochondrial defects and promotes survival in EV-A71 infected MNs
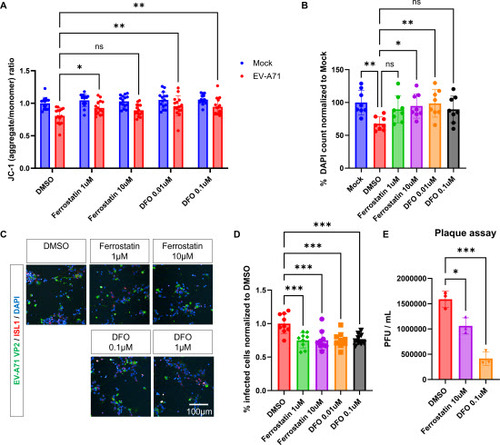
Having established that EV-A71-infected motor neurons undergo ferroptosis, we were motivated to investigate whether ferroptosis inhibition would mitigate EV-A71-induced cell death. Towards this end, we infected hESC-derived neurons with EV-A71, and treated them with the ferroptosis inhibitor ferrostatin-1, the free iron chelator deferoxamine (DFO) or DMSO control for at least 1 h before addition of the virus, followed by post-infection treatment for 24 h or 48 h. We found that treatment with ferrostatin or DFO resulted in partial restoration of MMP, suggesting that the small molecules improved mitochondrial function in neurons infected with EV-A71 (A). More importantly, treatment of infected neurons with ferrostatin-1 or DFO led to a significant increase in the number of viable cells by 32 - 46 % compared to the DMSO control, providing clear evidence that these small molecules effectively mitigate cell death following EV-A71 infection (B & S15). Expanding on this result, we observed a significant 23 - 25 % reduction in EV-A71 VP2 signal in EV-A71-infected neurons treated with ferrostatin-1 or DFO (C & 5D). Furthermore, DFO in particular, also led to a significant 3.8 ± 1.2 -fold reduction of plaque forming units (PFUs) (E), suggesting that free label iron may be exploited by the virus to support its intracellular replication. Consequently, chelation of free labile iron rescued the neuronal phenotypes associated with EV-A71 infection. This further implies that ferroptosis inhibitors hold promise as a potential neuroprotection strategy following EV-A71 infection.
Figure 5. Ferroptosis inhibitors restore mitochondrial defects, promote survival, and decrease infection rates in EV-A71 infected MNs. A. Mitochondrial membrane potential in EV-A71-infected or mock cultures treated with ferroptosis inhibitors, ferrostatin-1 and deferoxamine (DFO) at 24 hpi. n=12, Two-way ANOVA. B. DAPI counts at 48 hpi normalized to mock-infected control. n=12, One-way ANOVA. C. Immunodetection of VP2 in MNs treated with ferroptosis inhibitors at 48 hpi. D. Percentage of VP2+ cells. n=9, One-way ANOVA. E. Viral titers determined by plaque assay in culture supernatants collected at 48 hpi. n=3, One-way ANOVA. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
Discussion
EV-A71 is a neurotropic virus that infects motor neurons at neuromuscular junctions to invade the central nervous system [26]. We report here a novel, highly relevant culture system using human iPSC to study EV-A71 neuropathogenesis. Previous studies have used the neuroblastoma cell line SH-SY5Y to study EV-A71 neurovirulence. However, the main issues are that 1) neurons derived from SH-SY5Y respond to external stimuli (e.g. cellular stress, viral infections) differently from specialized neurons such as motor neurons, and 2) the SH-SY5Y cultures are homogeneously made up of only one cell type and are not representative of in vivo systems. Although more physiologically-relevant models, such as mouse motor neuron-like NSC-34 cells [17,18,26] and primary motor neurons from hSCARB2-transgenic mice cultured in microfluidic devices [5] have been recently used investigate the host response of motor neurons during EV71 infections, these models are murine-based and may not fully recapitulate human disease. In this study, by using a combination of a heterogeneous population of human spinal neurons derived from pluripotent stem cells and single cell transcriptomics, we discovered that EV-A71 preferentially infects and replicates in motor neurons. High viral load in human motor neurons then triggered the downregulation of ferritin, resulting in ferroptosis.
The advantage of employing scRNA-Seq in the heterogeneous human neuronal cultures lies in its ability to discern differences in viral loads between individual cells and populations, identify changes in gene expression between cells with high versus low viral loads, and explore both common and cell-specific events. This led to our discovery that human motor neurons exhibited higher EV-A71 viral load, despite constituting just 5% of the entire culture. Amongst the spinal neurons we detected in the hESC-derived heterogeneous culture, we found that more than 50% of the motor neurons have high viral load, compared to 30% of V1 and V2a interneurons and less than 10% of V2b interneurons. These heterogeneities in viral loads could suggest differences in virus tropism and/or replication dynamics in motor neurons versus other neurons. In our study, we found that motor neurons expressed higher levels of the host factor SCARB2 compared to other spinal neurons and neural progenitors (J-K & S15), which may partially explain why motor neurons are more permissive to EV-A71 infection. Likewise, higher expression of ANXA2 in motor neurons also promotes the replication of EV-A71 by facilitation the formation of the EV-A71 RNA replication complex [27]. The scRNA-Seq dataset also identifies unique genes that are highly expressed in MNs and show significant changes post-infection, potentially providing new insights into MN tropism but will require further validation (Figure S16).
In terms of the neurovirulence of EV-A71, we have identified mitochondrial dysfunctions and ferroptosis as pathways that could contribute to neuronal dysfunction and death, which sometimes result in complications such as paralysis. This finding is consistent with our earlier observations that EV-A71-infected murine motor neuron-like NC34 cells displayed impaired mitochondrial function [18]. Furthermore, recent studies have shown that EV-A71 leads to increased mitochondrial reactive oxygen species (ROS) and reduced MMP [28,29]. Expanding on this, we evaluated if elamipretide (SS31), a peptide compound that targets and stabilizes cardiolipin to improve mitochondrial function, would rescue EV-A71-induced neuronal defects. While SS31 treatment partially restored MMP, it was not sufficient to rescue the cells from EV-A71-induced cell death. On the other hand, we found that treatment of EV-A71-infected neurons with 10 nM DFO resulted in improved motor neuron survival, reduced infectivity as well as improved MMP, suggesting iron dyshomeostasis acts upstream of mitochondrial dysfunction in EV-A71 infected neurons.
Our study has also highlighted the differential host responses to EV-A71 in various neural subtypes. This is exemplified by the downregulation of FTL and FTH1 which is most evident in motor neurons or other neurons and less so in early neurons or neural progenitors (A). Both FTL and FTH1 are known transcriptional targets of the transcription factor Nrf2 [30], while Nrf2 nuclear translocation has been shown to decrease in mouse cells following EV-A71 infection [31]. Taken together, our results complement an expanding body of evidence which characterizes the anti-ferroptotic role of Nrf2 by preventing iron accumulation through regulation of ferritin expression. Since Nrf2-regulated ferroptosis has been indicated in neurodegenerative diseases of the motor neurons [32-34], it would be interesting to investigate whether this provides a linkage between enterovirus infection and neurodegenerative diseases that have been suggested previously [35,36].
Although ferroptosis has been reported in enterovirus-induced cell death and other virus-induced cell death [37-40], it is previously unknown if ferroptosis is associated with EV-A71 infection in human motor neurons. Here, we report multiple evidence strongly suggesting that EV-A71 infection results in high levels of intracellular iron through the concerted action of elevated TFRC and reduction of ferritin, ultimately causing cell death via ferroptosis. Consequently, the use of the iron chelator, DFO, proved to be more effective than the downstream ferroptosis inhibitor, Fer-1.
Previous studies have shown that EV-A71-infected RD cells also undergo apoptosis or pyroptosis in addition to ferroptosis [40], and we sought to understand the significance of ferroptosis in our study. In EV-A71-infected human motor neurons, we also observed activation of the apoptosis pathway but only marginal increase in the pyroptosis and necroptosis markers (Figure S10, S11, S17). While further studies will be needed to understand the exact mechanisms by which EV-A71 infection regulates these various cell death processes, our results nevertheless elucidated the role of ferritin and iron homeostasis in human motor neuron death after EV-A71 infection, highlighting iron chelation as a viable antiviral and neuroprotection strategy. We also conducted a preliminary screen for some of the more novel iron chelators that are commercially available and found that deferasirox may also reduce infection rate and cell death in the EV-A71-infected human motor neurons (Figure S18). While some of these ferroptosis inhibitors and iron chelators have been used clinically for the treatment of iron overload [41], the efficacy of the novel iron chelators still needs to be studied in more detail, and caution should be exercised, especially regarding the safety of these drugs in children.
In conclusion, we have demonstrated a highly relevant human spinal neuron model that allowed us to investigate the heterogeneity in host response to EV-A71 infection. Amongst the spinal neurons and progenitor cells within the culture, we found that motor neurons were most permissive to EV-A71 infection, which was explained by their intrinsically high expression of SCARB2 and AXNA2. We also found that EV-A71-infected motor neurons exhibited mitochondrial dysfunction with elevated labile Fe2+, peroxidated lipids, and increased cell death, which could be rescued by DFO, suggesting that reduction of the labile iron pool could be an effective neuroprotection strategy against neurological complications caused by EV-A71.
SYN_supplementary_EMI_revised_clean version.docx
Download MS Word (20.3 MB)DEGs in all neural cell ppopulations.zip
Download Zip (452 KB)Acknowledgements
We thank the flow cytometry unit at the Institute of Molecular and Cell Biology (IMCB) for technical support. We also thank the Central Imaging facility (IMCB) and the SBIC-Nikon Imaging Centre at Biopolis for assistance with microscopy. This work is supported by the National Research Foundation Singapore (NRF-CRP21-2018-0004) and IMCB, Agency for Science, Technology and Research.
Disclosure statement
The authors have no competing interests to declare.
References
- Zhang Q, MacDonald NE, Smith JC, et al. Severe enterovirus type 71 nervous system infections in children in the Shanghai region of China: clinical manifestations and implications for prevention and care. Pediatr Infect Dis J. 2014 May;33(5):482-7.
- Teoh HL, Mohammad SS, Britton PN, et al. Clinical Characteristics and Functional Motor Outcomes of Enterovirus 71 Neurological Disease in Children. JAMA Neurol. 2016 Mar;73(3):300-7.
- Muehlenbachs A, Bhatnagar J, Zaki SR. Tissue tropism, pathology and pathogenesis of enterovirus infection. J Pathol. 2015 Jan;235(2):217-28.
- Chen CS, Yao YC, Lin SC, et al. Retrograde axonal transport: a major transmission route of enterovirus 71 in mice. J Virol. 2007 Sep;81(17):8996-9003.
- Ohka S, Hao Tan S, Kaneda S, et al. Retrograde axonal transport of poliovirus and EV71 in motor neurons. Biochem Biophys Res Commun. 2022 Oct 20;626:72-78.
- Wang YF, Yu CK. Animal models of enterovirus 71 infection: applications and limitations. J Biomed Sci. 2014 Apr 17;21(1):31.
- Lin YW, Yu SL, Shao HY, et al. Human SCARB2 transgenic mice as an infectious animal model for enterovirus 71. PLoS One. 2013;8(2):e57591.
- Yamayoshi S, Koike S. Identification of a human SCARB2 region that is important for enterovirus 71 binding and infection. J Virol. 2011 May;85(10):4937-46.
- Yap MS, Tang YQ, Yeo Y, et al. Pluripotent Human embryonic stem cell derived neural lineages for in vitro modelling of enterovirus 71 infection and therapy. Virol J. 2016 Jan 6;13:5.
- Singh S, Poh CL, Chow VT. Complete sequence analyses of enterovirus 71 strains from fatal and non-fatal cases of the hand, foot and mouth disease outbreak in Singapore (2000). Microbiol Immunol. 2002;46(11):801-8.
- Brown BA, Pallansch MA. Complete nucleotide sequence of enterovirus 71 is distinct from poliovirus. Virus Res. 1995 Dec;39(2-3):195-205.
- Wu Y, Yeo A, Phoon MC, et al. The largest outbreak of hand; foot and mouth disease in Singapore in 2008: the role of enterovirus 71 and coxsackievirus A strains. Int J Infect Dis. 2010 Dec;14(12):e1076-81.
- Hor JH, Santosa MM, Lim VJW, et al. ALS motor neurons exhibit hallmark metabolic defects that are rescued by SIRT3 activation. Cell Death Differ. 2021 Apr;28(4):1379-1397.
- Hor JH, Soh ES, Tan LY, et al. Cell cycle inhibitors protect motor neurons in an organoid model of Spinal Muscular Atrophy. Cell Death Dis. 2018 Oct 27;9(11):1100.
- Ng S-Y, Soh Boon S, Rodriguez-Muela N, et al. Genome-wide RNA-Seq of Human Motor Neurons Implicates Selective ER Stress Activation in Spinal Muscular Atrophy. Cell Stem Cell. 2015 2015/11/05/;17(5):569-584.
- Chooi WH, Ng CY, Ow V, et al. Defined Alginate Hydrogels Support Spinal Cord Organoid Derivation, Maturation, and Modeling of Spinal Cord Diseases. Adv Healthc Mater. 2023 Apr;12(9):e2202342.
- Too IH, Yeo H, Sessions OM, et al. Enterovirus 71 infection of motor neuron-like NSC-34 cells undergoes a non-lytic exit pathway. Sci Rep. 2016 Nov 16;6:36983.
- Too IHK, Bonne I, Tan EL, et al. Prohibitin plays a critical role in Enterovirus 71 neuropathogenesis. PLoS Pathog. 2018 Jan;14(1):e1006778.
- Li D, Su M, Sun PP, et al. Global profiling of the alternative splicing landscape reveals transcriptomic diversity during the early phase of enterovirus 71 infection. Virology. 2020 Sep;548:213-225.
- Birk AV, Liu S, Soong Y, et al. The mitochondrial-targeted compound SS-31 re-energizes ischemic mitochondria by interacting with cardiolipin. J Am Soc Nephrol. 2013 Jul;24(8):1250-61.
- Omiya S, Hikoso S, Imanishi Y, et al. Downregulation of ferritin heavy chain increases labile iron pool, oxidative stress and cell death in cardiomyocytes. J Mol Cell Cardiol. 2009 Jan;46(1):59-66.
- Ou M, Jiang Y, Ji Y, et al. Role and mechanism of ferroptosis in neurological diseases. Mol Metab. 2022 Jul;61:101502.
- Chen X, Yu C, Kang R, et al. Iron Metabolism in Ferroptosis. Front Cell Dev Biol. 2020;8:590226.
- Yi L, Hu Y, Wu Z, et al. TFRC upregulation promotes ferroptosis in CVB3 infection via nucleus recruitment of Sp1. Cell Death Dis. 2022 Jul 11;13(7):592.
- Yi L, Yang Y, Hu Y, et al. Complement components regulates ferroptosis in CVB3 viral myocarditis by interatction with TFRC. Free Radic Biol Med. 2024 Feb 20;212:349-359.
- Lim ZQ, Ng QY, Oo Y, et al. Enterovirus-A71 exploits peripherin and Rac1 to invade the central nervous system. EMBO Rep. 2021 Jun 4;22(6):e51777.
- Zhang Q, Li S, Lei P, et al. ANXA2 Facilitates Enterovirus 71 Infection by Interacting with 3D Polymerase and PI4KB to Assist the Assembly of Replication Organelles. Virol Sin. 2021 Dec;36(6):1387-1399.
- Zhang W, Yang H, Liu Z, et al. Enterovirus 71 leads to abnormal mitochondrial dynamics in human neuroblastoma SK-N-SH cells. Virus Res. 2024 Jan 2;339:199267.
- Foo J, Bellot G, Pervaiz S, et al. Mitochondria-mediated oxidative stress during viral infection. Trends Microbiol. 2022 Jul;30(7):679-692.
- He J, Abikoye AM, McLaughlin BP, et al. Reprogramming of iron metabolism confers ferroptosis resistance in ECM-detached cells. iScience. 2023 Jun 16;26(6):106827.
- Cui G, Wang H, Yang C, et al. Berberine prevents lethal EV71 neurological infection in newborn mice. Front Pharmacol. 2022;13:1027566.
- Abdalkader M, Lampinen R, Kanninen KM, et al. Targeting Nrf2 to Suppress Ferroptosis and Mitochondrial Dysfunction in Neurodegeneration. Front Neurosci. 2018;12:466.
- Yang B, Pan J, Zhang XN, et al. NRF2 activation suppresses motor neuron ferroptosis induced by the SOD1(G93A) mutation and exerts neuroprotection in amyotrophic lateral sclerosis. Neurobiol Dis. 2023 Aug;184:106210.
- Xiang Y, Song X, Long D. Ferroptosis regulation through Nrf2 and implications for neurodegenerative diseases. Arch Toxicol. 2024 Mar;98(3):579-615.
- Xue YC, Feuer R, Cashman N, et al. Enteroviral Infection: The Forgotten Link to Amyotrophic Lateral Sclerosis? Front Mol Neurosci. 2018;11:63.
- Swanson NR, Fox SA, Mastaglia FL. Enterovirus hypothesis for motor neurone disease. BMJ. 1994 Sep 17;309(6956):743.
- Xiao L, Huang H, Fan S, et al. Ferroptosis: A mixed blessing for infectious diseases. Front Pharmacol. 2022;13:992734.
- Wang J, Zhu J, Ren S, et al. The role of ferroptosis in virus infections [Review]. Frontiers in Microbiology. 2023 2023-November-23;14.
- Wang MP, Joshua B, Jin NY, et al. Ferroptosis in viral infection: the unexplored possibility. Acta Pharmacol Sin. 2022 Aug;43(8):1905-1915.
- Kung YA, Chiang HJ, Li ML, et al. Acyl-Coenzyme A Synthetase Long-Chain Family Member 4 Is Involved in Viral Replication Organelle Formation and Facilitates Virus Replication via Ferroptosis. mBio. 2022 Feb 22;13(1):e0271721.
- Entezari S, Haghi SM, Norouzkhani N, et al. Iron Chelators in Treatment of Iron Overload. J Toxicol. 2022;2022:4911205.