
ABSTRACT
The most common and lethal type of lung cancers with close to 1.8 million cases and less than 20% survival rate after diagnosis was the NSCLC. It also accounted for about 80% to 85% of the reported lung cancer cases. To combat this problem, more effective NSCLC drugs are needed. Molecular docking-based virtual screening was performed to provide a firsthand information on the binding modes between the AZD9291 derivatives and the EGFR receptor. The docking-based virtual screening has identified six compounds (17, 12, 15 1, 10 and 19) as the hit with compound 17 as the best hit with a mole dock score of -151.71 kcal/mol, respectively. It interacted with these group of amino acids ASP800 (2.26Å), PRO794 (2.87Å), MET793 (2.29Å) and CYS797 (2.65Å) using hydrogen bond, and with LEU799, ARG841(4), PHE723, LEU718 and CYS797 group of amino acids using hydrophobic interaction, respectively. All the hit compounds recognized were realized to have better mole dock scores than the Osimertinib (AZD9291) with a mole dock score of -118.87 kcal/mol. The hit compounds were ascertained to be drug-like and possessed good pharmacokinetic profiles. The value of their bioavailability score (0.55) further confirmed that they are orally bioavailable.
Introduction
Lung cancer remains the foremost cause of tumor-related death globally. This tumor (lung cancer) is divided into oat cell lung cancer (small-cell lung cancer, SCLC) and non–small cell lung cancer (NSCLC), respectively [Citation1]. NSCLC, which comprises of bulky cell carcinoma, glandular carcinoma and squamous cell carcinoma, was the common and lethal type of lung cancer with nearly 1.8 million cases and about 20% rate of survival in every 5 years after diagnosis at all stages and 2.0% rate of survival in every 5 years at the fourth (IV) stage [Citation2,Citation3]. NSCLC accounted for about 80–85% of the reported lung cancer cases, while the oat cell lung cancer accounted for about 15 to 20% remaining lung cancer cases [Citation4].
Epidermal growth factor receptor (EGFR) was discovered in the University of Vanderbilt (USA) by a scientist named Stanley Cohen in 1956 [Citation5]. EGFR is a member of the receptor tyrosine kinase (RTK) and belongs to the ErbB family of proteins that regulate essential cellular roles, such as cell division, growth, differentiation, metabolism, adhesion, motility and death [Citation6]. It was recognized as the principal target for the treatment of NSCLC [Citation7].
Erlotinib and Gefitinib, the FDA-approved first-generation EGFR tyrosine kinase inhibitors (TKIs), provide substantial medical assistance in patients with NSCLC by harboring the EGFR activating (L858R) and classical (del. E746-A750) mutations in exon 19 and exon 21, respectively [Citation8]. Furthermore, patients harboring L858R EGFR mutations usually respond well to gefitinib and erlotinib, the so-called first-generation EGFR-TKIs. However, the time frame for their efficacy was very limited due to the development of resistance by the EGFRT790M (the so-called gatekeeper mutation), which was observed in about 50% of patients with such EGFR mutation (EGFRT790M) [Citation9].
Dacomitinib and Afatinib, the FDA-approved irreversible second-generation EGFR-TKIs capable of covalently alkylating the cysteine 797 residue near the EGFR ATP binding site, were developed to resolve the induced EGFRT790M mutation-related resistance to the so-called first-generation EGFR-TKIs [Citation8,Citation10]. Unfortunately, their clinical effectiveness was limited due to serious side effects such as skin rash and gastrointestinal toxicity, as well as a lack of selectivity between EGFR mutant and wild-type EGFR (EGFRWT) [Citation10,Citation11].
Rociletinib (CO-1686) and Osimertinib (AZD9291), the FDA-approved irreversible third-generation EGFR-TKIs, were designed and developed in order to inhibit the EGFRT790M resistance mutation (to first-generation EGFR-TKIs) while being more selective to the EGFRWT and also to overcome the observed toxicity of the irreversible second-generation EGFR-TKIs [Citation10,Citation12,Citation13].
This study aimed at carrying out theoretical validation of some potential third-generation EGFR-TKIs as NSCLC drugs using molecular docking-based virtual screening, drug-like and ADMET feature predictions.
Materials and methods
Molecular docking virtual screening methodology
Sourcing of dataset
Zhao et al. synthesized and reported a set of third-generation epidermal growth factor receptor (EGFR)L858R/T790M inhibitors (AZD9291 derivatives) as NSCLC drugs [Citation14]. This set of compounds was evaluated for their EGFR inhibitory activities against EGFRWT and EGFRT790M/L858R EGFR kinases, respectively. For the purpose of this study, EGFR inhibitory activities against EGFRT790M/L858R EGFR kinases were selected and used in order to carry out theoretical validation of their enzymatic inhibitory activities against EGFRT790M/L858R kinase. presents the canonical SMILES of the selected compounds with the mole dock score in kcal/mol against the EGFRT790M/L858R kinase.
Table 1. The canonical SMILES AZD9291 derivatives.
Drawing of structure of the sourced dataset and optimization
The software developed by the University of Cambridge (Chemdraw) was used in drawing the two-dimensional structures of the sourced dataset [Citation15]. In this work, the Merck molecular force field (MMFF) with density functional theory (DFT) at B3LYP/6-311 G* theory level (Becke’s three-parameter hybrid function utilizing LYP correlation functional using 6-311 G* basis set) was used for the optimum conformational search for all the 27 sets of 2,4-diaminopyrimidine derivatives [Citation10,Citation16].
Library of compounds preparation
The optimum conformations obtained after geometry optimization of the 27 sets of 2,4-diaminopyrimidine derivatives were saved in a protein data bank file format. The 27 sets of 2,4-diaminopyrimidine derivatives were further prepared using the default setting of the Molegro virtual docker (MVD) by selecting ‘if missing’ to the following parameters: assign bonds, assign bond orders and hybridization, assign charges and assign tripos atom types, as well as creating explicit hydrogens and detecting flexible torsions, respectively [Citation17,Citation18].
Identification of the active site
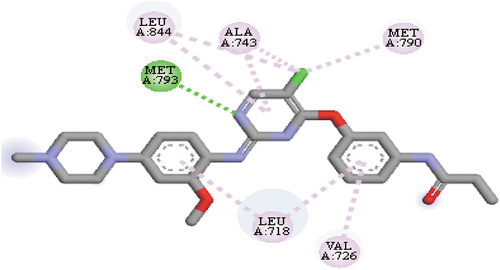
The EGFRL858R/T790M receptor crystal structure and its co-crystalized/native ligand (WZ4002) with a protein data bank (pdb) entry identifier: 3IKA was retrieved from the RCSB pdb database (https://www.rcsb.org/) [Citation19]. In this work, for the identification of the group of amino acids in the binding pose/active site of the EGFRL858R/T790M receptor, the native ligand fused to the EGFRL858R/T790M receptor was visualized with version 16.1.0.15350 of discovery studio. The group of amino acids identified in the binding pose/active site of the EGFRL858R/T790M receptor were ALA743, LEU844, LEU718, MET793, MET790 and VAL726, respectively. shows the 2D view of the native ligand (WZ4002) in the binding pose/active site of the EGFRL858R/T790M receptor.
Figure 1. The native ligand (WZ4002) in the binding pose/active site of EGFRL858R/T790M receptor.
EGFR receptor preparation
Before carrying out the molecular docking investigation, the EGFR receptor was imported on to the MVD 3D view space (interface), and then residues with structural errors were rebuilt and repaired. In the process of preparing the EGFR receptor, the surface was created and cavities were detected (by right-clicking on the EGFR receptor and clicking surface and detecting cavities) before removing the native ligand from the EGFR receptor, respectively.
Investigation of the molecular docking-based virtual screening
Molegro Virtual Docker (MVD) was used to perform the protein-ligand docking investigation in this research due to its high accuracy as compared to other docking software. The execution of the docking process/protocol was done by selecting plant score as the docking algorithm (scoring function) and defining the binding site as a spherical region that consists of all the protein cavities within 24 Å. For all other calculations, default settings were sustained and preserved. The docking was performed using a grid resolution of 0.30 Å and for each of the 10 independent runs; a maximum number of 1500 iterations were executed on a single population of 50 individuals [Citation20].
Validation of the molecular docking protocol
The validation of the molecular docking protocol was carried out by re-docking WZ4002 into the active sites of 3IKA receptor. The residues of the re-docked co-crystalized ligands were then compared to those of the investigated compounds in this study.
Modeling of the drug-likeness and ADMET features
The modeling of the drug-likeness and ADMET features of these investigated compounds were performed using the SWISSADME (http://www.swissadme.ch/index.php) and pkCSM (http://structure.bioc.cam.ac.uk/pkcsm) online web servers, respectively [Citation15,Citation21,Citation22].
Results and discussion
Molecular docking virtual screening
MVD was used to carry out the molecular docking virtual screening to investigate the ligand–protein interactions in this research and also to provide firsthand information on the binding modes of the investigated compounds and the ATP-binding pocket of the EGFR receptor. The molecular docking virtual screening was performed between AZD9291 derivatives and EGFR receptor with the protein data bank (pdb) entry identifier: 3IKA, respectively. The mole dock score values were used to rank the investigated compounds for their binding affinities in the ATP-binding pocket of the EGFR receptor. The range at which the mole dock score values of the investigated compounds were found between −116.66 to −151.71 kcal/mol, respectively (). The results of the best top 6 will be presented and discussed ().
Table 2. Mode of interactions of the best six compounds and the EGFR receptor.
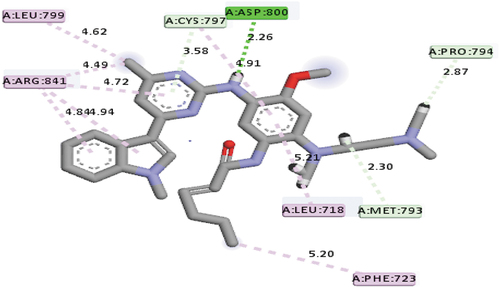
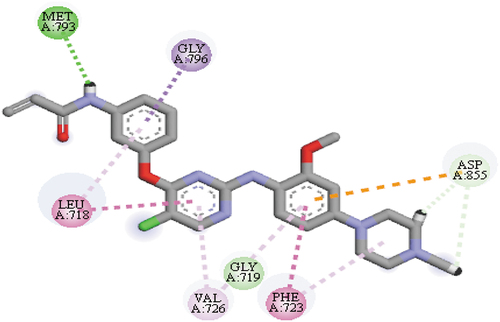
The molecular docking-based virtual screening executed on the AZD9291 derivatives have recognized compound 17 with a mole dock score of −151.71 kcal/mol as the top hit among the investigated compounds. Hydrogen bond interactions at different bond distances with these group of amino acids ASP800 (2.26 Å), PRO794 (2.87 Å), MET793 (2.29 Å) and CYS797 (2.65 Å) were observed as among the type interactions between compound 17 and the ATP-binding pocket of the EGFR receptor. Apart from hydrogen bond interactions, hydrophobic interaction was also observed between compound 17 and the ATP-binding pocket of the EGFR receptor with LEU799, ARG841(4), PHE723, LEU718 and CYS797 group of amino acids, respectively. Based on this information, it can be seen that the compound interacted with the most important group of amino acids MET793 and CYS797 via hydrogen bond interactions, which indicated the possibility of the compound to manage the resistance caused by the EGFRL858R/T790M mutant. depicts the mode of interactions between compound 17 and the binding pocket of EGFR receptor.
Figure 2. Mode of interactions between compound 17 and EGFR receptor.
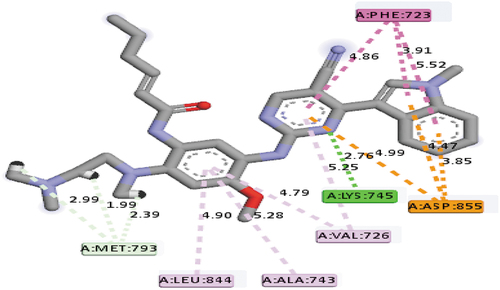
The second top best compound recognized in the series of the compound investigated was compound 12 with a mole dock score of −146.77 kcal/mol, respectively. The following group of amino acids: LYS745 (2.76 Å), MET793 (2.99 Å), MET793 (1.99 Å), and MET793 (2.39 Å) in the ATP-binding pocket of the EGFR receptor were seen to interact with the compound using hydrogen bonds at the specified distances. It also formed a hydrophobic interaction with the binding pocket of the EGFR receptor with these group of amino acids: PHE723 (3), VAL726 (2), ALA743 & LEU844, respectively. Electrostatic interaction was also observed between it and the binding pocket of the receptor with the ASP855 amino acid, respectively. This compound can also serve as a potential EGFR inhibitor since it interacts with a vital group of amino acid, MET793, via hydrogen bond, which might cause the management of the resistance caused by the EGFRL858R/T790M mutant. depicts the mode of interaction between compound 12 and the binding pocket of the EGFR receptor.
Figure 3. Mode of interactions between compound 12 and EGFR receptor.
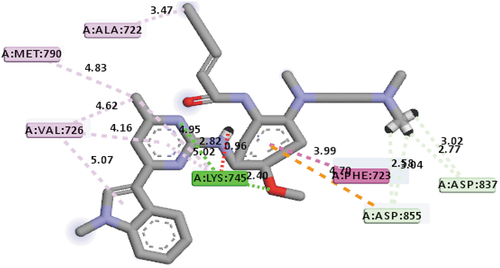
The third best hit compound recognized was compound 15, which has a mole dock score value of −145.40 kcal/mol. The mode of interactions between it and binding pocket of the EGFR receptor was using hydrogen bond with these group of amino acids at bond distances indicated: LYS745 (2.82Å), LYS745 (2.40Å), ASP837 (3.02Å), ASP837 (2.77Å), ASP855 (3.04Å) and ASP855 (2.58Å), hydrophobic interactions with these group of amino acids: PHE723, ALA722, VAL726 (3), LYS745 (2) and MET790, and electrostatic interaction with ASP855 group of amino acid, respectively. As such, compound 12 can also serve as an EGFR inhibitor because of its interaction with the MET790 group of amino acids, respectively. depicts the mode of interactions between compound 15 and EGFR receptor.
Figure 4. Mode of interactions between compound 15 and EGFR receptor.
The fourth best hit compounds recognized were compounds 1 and 10, each with a mole dock score of −144.10 and −144.09 kcal/mol, respectively. The mode of interactions between compound 1 and the binding pocket of the EGFR receptor was through hydrogen bonds with these group of amino acids: THR854 (2.30Å), ASP855 (2.73Å), THR854 (2.09Å) and GLN791 (2.31Å), hydrophobic with these group of amino acids: LEU718, GLY796, MET790, LEU718, ALA743, MET793, LEU844, VAL726 and LYS745, and other interactions as identified by the software with this group of amino acids MET790, respectively. For compound 10 and the binding pocket of the EGFR receptor, the mode of interactions was also via hydrogen bonding with PRO794 (2.16Å) and MET793(2.18Å), hydrophobic with PHE723 (2), ARG841, VAL726 (2), LEU718 and ALA743, electrostatic with ASP855 group of amino acids, respectively. These two compounds (i.e. compounds 1 and 10) can also serve as EGFR inhibitors because of its interaction with MET790 and MET793 group of amino acids, respectively. depict the mode of interactions between compounds 1 and 10 and EGFR receptor.
Figure 5. Mode of interactions between compound 1 and EGFR receptor.
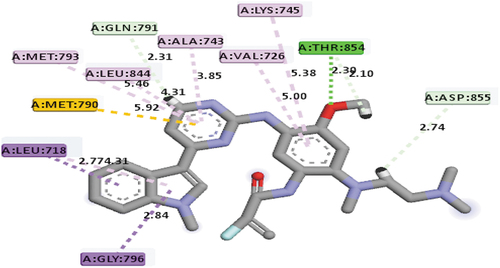
Figure 6. Mode of interactions between compound 10 and EGFR receptor.
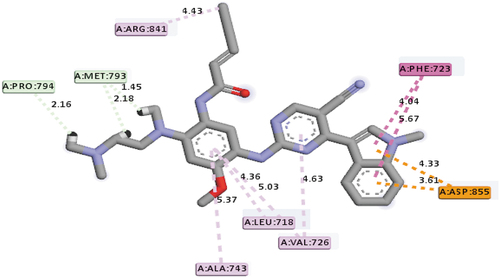
The fifth, which is the last among the top-hit compounds recognized in this study, was compound 19 with a mole dock score of −143.26 kcal/mol, respectively. Hydrogen bond interactions at different bond distances with these group of amino acids ASP800 (2.49Å), PHE795 (2.45Å), ASP800 (2.50Å) and MET793 (2.49Å) were observed as among the types of interactions between compound 19 and the ATP-binding pocket of the EGFR receptor. Apart from hydrogen bond interactions, hydrophobic interaction was also observed between compound 19 and the ATP-binding pocket of the EGFR receptor with VAL726 (2), ALA743, LYS745 and LEU718 group of amino acids, respectively. On the other hand, electrostatic interaction was also observed with the MET790 amino acid residue. Based on the mode of interaction between it and the EGFR receptor with MET793 and MET790 group of amino acids, it indicated the possibility of the compound to manage the resistance caused by the EGFRL858R/T790M mutant. depicts the mode of interactions between compound 19 and the binding pocket of EGFR receptor.
Figure 7. Mode of interactions between compound 19 and EGFR receptor.
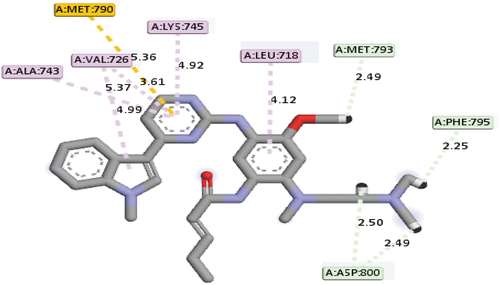
These group of amino acids were virtually common to almost all the recognized top best hit compounds and Osimertinib (AZD9291): MET793, MET790, ALA743, LEU718, LEU844, VAL726, LYS745, ASP855 and PHE723, respectively.
Osimertinib (AZD9291), a third-generation FDA-approved drug for the treatment of EGFR mutation, was also docked into the active site of the EGFR receptor in order to make a comparison between the standard drug (AZD9291) and the identified best-hit compounds. AZD9291 with a mole dock score of −118.87 kcal/mol interacted with the following group amino acids ASP855 (2.11Å), LYS745 (2.55Å), ASP855 (2.64Å) and PRO794 (2.33Å) via hydrogen bond, and LEU718(3), LEU844, VAL726 and LEU792 via hydrophobic, respectively (). All the identified hit compounds were observed to have better mole dock scores than the standard drug (AZD9291).
Figure 8. Mode of interactions between AZD9291 and EGFR receptor.
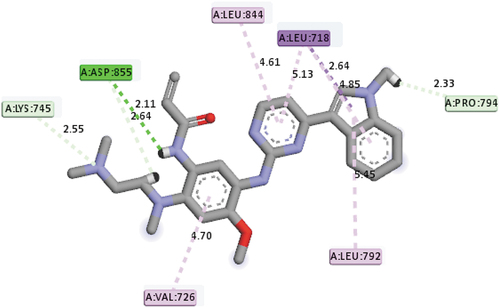
In order to successfully validate the molecular docking procedure, WZ4002 was then re-docked into the active site of the EGFR receptor. WZ4002 with a mole dock score of −103.728 kcal/mol interacted with the following amino acid residues: MET793, ASP855, GLY796, PHE723, LEU718, GLY719, VAL726 after successfully re-docking it into the active site of the enzyme (). The following group of amino acids ASP855, MET793, LEU718, VAL726 and PHE723 were common to the best hit compounds, Osimertinib (AZD9291) and the WZ4002, respectively. This further established the high accuracy of the molecular docking method used.
Figure 9. Mode of interactions between WZ4002 and EGFR receptor.
According the information retrieved from this investigation, it provided a strong theoretical validation that the investigated compounds can inhibit the EGFRL858R/T790M mutations. This further serves as an evidence that the investigated compounds can be used as potential third generation EGFR inhibitors after passing the pre-clinical trials.
Drug-like/admet prediction and evaluation
The drug-like features of the hit compounds were assessed adopting the prominent Lipinski’s rule of five (Molecular weight≤500, Number of hydrogen bond donors≤5, Number of hydrogen bond acceptors≤10, and Calculated MLog p ≤ 5) using SwissADME web server (). The modeled drug-like features showed only 1 violation for all the reported compounds (they all have molecular weight greater than 500). Their number of hydrogen bond donors, number of hydrogen bond acceptors and the calculated MLog p values were all within the accepted range. Based on this screening conditions, the recognized hit compounds were ascertained to be drug-like (druggable) by not violating more than the threshold values set for the Lipinski’s rule of five. Furthermore, according to the value of their bioavailability score (0.55), they were all further established to be orally bioavailable. Generally, these recognized hit compounds were predicted not to have any issue associated to their bioavailability.
Table 3. Drug-like features of the best hit compounds using Lipinski’s RO5 screening conditions.
The modeled ADMET features of the hit compounds are presented in . They were all observed to have human intestinal absorption between 90.635 to 93.573%, respectively. The values of their human intestinal absorption have passed the threshold accepted value of 30% set for the evaluation of this feature/property, this is an indication that these the hit compounds can be absorbed within the human intestine. The threshold accepted value set for BBB (that is the blood brain barrier) permeability is>0.3 to < −1 and CNS (that is central nervous system) permeability is > −2 to < −3, respectively. The BBB permeability for these the hit compounds were observed to be < −1 which simply mean that the compounds can better infiltrate/penetrate via the BBB, respectively. For their CNS permeability value, it is found to be > −2 but < −3 for all the hit compounds which indicated that they can better infiltrate/penetrate the CNS, respectively. The evaluation of a drug’s metabolism can make available key information about its pharmacokinetic and pharmacodynamic features. Generally, drugs are metabolized by a different group of enzymes which were identified as cytochrome P450 (CYP) enzymes. CYP3A4 is the most widely used among the cytochrome P450 (CYP) enzymes which described for approximately 30–40% of the total CYP content in human adult liver and small intestine (a good small molecule is expected to be a substrate and also inhibitor of CYP3A4). The hit compounds recognized were all observed to be both inhibitors and substrate of CYP3A4 class of enzymes. This is further established that these hit compounds can be breakdown in the body. Another significant feature is how these hit compounds can be removed from the body (excretion/total clearance), excretion/total clearance expresses the connection/relationship between the rate of their removal and concentration within the body. The best identified hit compounds displayed average values of total clearance but within the acknowledged threshold limit of a small molecule in the body. According to the toxicity test predicted, they were all detected to be nontoxic. Finally, the general ADMET features of these best hit compounds displayed their good pharmacokinetic profiles ().
Table 4. ADMET features of the best hit compounds.
Conclusion
Molecular docking-based virtual screening was performed to provide a firsthand information on the binding modes of the investigated compounds and the ATP-binding pocket of the EGFR receptor. The top best hit compound was compound 17 with a mole dock score of −151.71 kcal/mol which interacted with these group of amino acids ASP800 (2.26Å), PRO794 (2.87Å), MET793 (2.29Å) and CYS797 (2.65Å) using hydrogen bond, and with LEU799, ARG841(4), PHE723, LEU718 and CYS797 group of amino acids using hydrophobic interaction, respectively. All the hit compounds recognized were seen to have better mole dock scores than the Osimertinib (AZD9291) with a mole dock score of −118.872 kcal/mol. The following group of amino acids ASP855, MET793, LEU718, VAL726 and PHE723 were common to the best hit compounds, Osimertinib (AZD9291) and the WZ4002, respectively. This further confirmed the high precision of the method used.
The hit compounds recognized were ascertained to be drug-like in nature by not violating more than the threshold values set for each of the screening condition used for the evaluation of their drug-like feature. The value of their bioavailability score of 0.55 further confirmed that they orally bioavailable. And their ADMET features displayed their good pharmacokinetic profiles.
According the information retrieved from this investigation, it provided a strong theoretical validation of the investigated compounds against EGFR mutations. It serves and evidence that the investigated compounds can be used as potential third generation EGFR inhibitors after passing the pre-clinical trials.
Acknowledgments
The authors sincerely acknowledged Tertiary Education Trust Fund (Tetfund) for sponsoring this research under the Tetfund IBR research project grant (identification number: TETF/DR&D/UNI/ZARIA/IBR/2020/VOL.1/52) as well as Ahmadu Bello University, Zaria for its technical support in the course of this research.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Bistrović A, Krstulović L, Harej A, et al. Design, synthesis and biological evaluation of novel benzimidazole amidines as potent multi-target inhibitors for the treatment of non-small cell lung cancer. Eur J Med Chem. 2018;143:1616–1634.
- Khuri F. Lung cancer and other pulmonary neoplasms. Goldman-Cecil Med. 2016;2:1303–1313.
- Tièche CC, Peng R-W, Dorn P, et al. Prolonged pemetrexed pretreatment augments persistence of cisplatin-induced DNA damage and eliminates resistant lung cancer stem-like cells associated with EMT. BMC Cancer. 2016;16:125.
- Ibrahim MT, Uzairu A, Shallangwa GA, et al. Molecular docking investigation and pharmacokinetic properties prediction of some anilinopyrimidines analogues as EGFRT790M tyrosine kinase inhibitors. Egypt J Basic Appl Sci. 2021;8(1):203–213.
- Sato JD, Okamoto T, Barnes D, et al. A tribute to Dr. Gordon Hisashi Sato (December 24, 1927–march 31, 2017). In vitro Cell Dev Biol Anim. 2018;54(3):177–193.
- Othman IM, Alamshany ZM, Tashkandi NY, et al. New pyrimidine and pyrazole-based compounds as potential EGFR inhibitors: synthesis, anticancer, antimicrobial evaluation and computational studies. Bioorg Chem. 2021;114:105078.
- Hanan EJ, Baumgardner M, Bryan MC, et al. 4-Aminoindazolyl-dihydrofuro [3, 4-d] pyrimidines as non-covalent inhibitors of mutant epidermal growth factor receptor tyrosine kinase. Bioorg Med Chem Lett. 2016;26(2):534–539.
- Song Z, Huang S, Yu H, et al. Synthesis and biological evaluation of morpholine-substituted diphenylpyrimidine derivatives (Mor-DPPYs) as potent EGFR T790M inhibitors with improved activity toward the gefitinib-resistant non-small cell lung cancers (NSCLC). Eur J Med Chem. 2017;133:329–339.
- Ibrahim MT, Uzairu A, Shallangwa GA, et al. Lead identification of some anti-cancer agents with prominent activity against non-small cell lung cancer (NSCLC) and structure-based design. Chem Afr. 2020;3(4):1023–1044.
- Pawara R, Ahmad I, Surana S, et al. Computational identification of 2, 4-disubstituted amino-pyrimidines as L858R/T790M-EGFR double mutant inhibitors using pharmacophore mapping, molecular docking, binding free energy calculation, DFT study and molecular dynamic simulation. In silico Pharmacol. 2021;9(1):1–22.
- Patel H, Pawara R, Ansari A, et al. Recent updates on third generation EGFR inhibitors and emergence of fourth generation EGFR inhibitors to combat C797S resistance. Eur J Med Chem. 2017;142:32–47.
- Chen L, Fu W, Feng C, et al. Structure-based design and synthesis of 2, 4-diaminopyrimidines as EGFR L858R/T790M selective inhibitors for NSCLC. Eur J Med Chem. 2017;140:510–527.
- Karnik KS, Sarkate AP, Tiwari SV, et al. Computational and synthetic approach with biological evaluation of substituted quinoline derivatives as small molecule L858R/T790M/C797S triple mutant EGFR inhibitors targeting resistance in non-small cell lung cancer (NSCLC). Bioorg Chem. 2021;107:104612.
- Zhao B, Xiao Z, Qi J, et al. Design, synthesis and biological evaluation of AZD9291 derivatives as selective and potent EGFRL858R/T790M inhibitors. Eur J Med Chem. 2019;163:367–380.
- Mills N. ChemDraw Ultra 10.0 CambridgeSoft, 100 CambridgePark Drive, Cambridge, MA 02140. www.cambridgesoft.com. Commercial Price: 1910fordownload, 2150 for CD-ROM; Academic Price: 710fordownload, 800 for CD-ROM. ACS Publications; 2006.
- Kohn W, Becke AD, Parr RG. Density functional theory of electronic structure. J Phys Chem. 1996;100(31):12974–12980.
- Ghamali M, Chtita S, Hmamouchi R, et al. The inhibitory activity of aldose reductase of flavonoid compounds: combining DFT and QSAR calculations. J Taibah Univ Sci. 2016;10(4):534–542.
- Ibrahim MT, Uzairu A, Shallangwa GA, et al. Structure-based design and activity modeling of novel epidermal growth factor receptor kinase inhibitors; an in silico approach. Sci Afr. 2020;e00503. DOI:10.1016/j.sciaf.2020.e00503
- Chang J, Ren H, Zhao M, et al. Development of a series of novel 4-anlinoquinazoline derivatives possessing quinazoline skeleton: design, synthesis, EGFR kinase inhibitory efficacy, and evaluation of anticancer activities in vitro. Eur J Med Chem. 2017;138:669–688.
- Abdullahi M, Uzairu A, Shallangwa GA, et al. Virtual molecular docking study of some novel carboxamide series as new anti-tubercular agents. Eur J Chem. 2020;11(1):30–36.
- Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7:42717.
- Hadni H, Elhallaoui M. 3D-QSAR, docking and ADMET properties of aurone analogues as antimalarial agents. Heliyon. 2020;6(4):e03580.