
Abstract
We describe a patient (CG) suffering from early onset dementia who presented with corticobasal syndrome (CBS). The aims of the study were as follows: (i) a detailed description of the cognitive phenotype; (ii) a comprehensive, longitudinal evaluation of apraxia; (iii) an appraisal of the impact of apraxia and other cognitive impairments on patient functional status; and (iv) an indirect mapping of degeneration spreading. A three-year longitudinal, observational follow-up study of cognitive and functional status was performed. Four main results emerged. First, an unusual CBS phenotype appeared that was characterized by symmetrical presentation, asymmetrical course, and prominent posterior (bi-parietal) cognitive and motor cortical manifestations. Second, some findings of limb apraxia in CBS were replicated and substantiated; moreover, some novel findings of other cognitive impairments emerged. Third, an early, significant functional decline, probably related to apraxia and to visuospatial attention impairments, became apparent. Fourth, CG’s clinical picture was compatible with an underlying dysfunction of the large-scale, dorsal sensory-motor association network, as already suggested in previous CBS cases. This case report confirms the heterogeneity of CBS and suggests the emergence of a possible less common variant, i.e. the posterior CBS (P-CBS).
Public Interest Statement
Our research group studies the dramatic mind’s decline resulting from the progressive brain damage caused by Alzheimer’s disease and other dementias. Specifically, we aim to precisely report all the impairments that dementia causes in mental abilities as well as in feelings and behavior of the patients, and how these disorders change over time. This scientific knowledge may help detect the early features of dementia to diagnose it as soon as possible. Moreover, we are interested in forecast what difficulties patients will have in their daily lives, as well as how they will behave in some circumstances and what emotions they will feel. The ultimate aim of this long-lasting and demanding study is to know better how to help patients and make them feel well. Also, we want to make patients’ mind stronger and to promote social engagement for them.
Competing interests
The authors declare no competing interest.
1. Introduction
Corticobasal syndrome (CBS) is a variable but recognizable neurological disorder with an insidious onset and a progressive course. It is characterized by an asymmetric, akinetic, and rigid syndrome, with prominent apraxia and other symptoms suggesting cortical (i.e. myoclonus, cortical sensory loss, alien limb phenomenon) and basal ganglionic dysfunction (e.g. dystonia, tremor) (Boeve, Lang, & Litvan, Citation2003; Burrell, Hodges, & Rowe, Citation2014; Graham, Bak, & Hodges, Citation2003; Mathew, Bak, & Hodges, Citation2011). Cognitive impairments and/or behavioral features are often associated with motor symptoms early on in the disease course (Burrell et al., Citation2014; Graham et al., Citation2003).
Apraxia is one of the core features in the current diagnostic criteria of CBS (Armstrong et al., Citation2013). In particular, limb apraxia is very frequent, with estimates between 70 and 80% of cases, and it is often a presenting feature (Stamenova, Roy, & Black, Citation2009). Many patients suffering from CBS also present with limb-kinetic apraxia; conversely, oro-facial apraxia may be absent or develop later in the disease progression (Ozsancak, Auzou, Dujardin, & Hannequin, Citation2000; Ozsancak, Auzou, & Hannequin, Citation2004; Stamenova et al., Citation2009). Finally, apraxia affecting other body parts (e.g. trunk, eyelid, gaze) has been reported less frequently (Okuda, Tanaka, Kawabata, Tachibana, & Sugita, Citation2001; Rajagopal, Bateman, & Van Stavern, Citation2011). Taking into account the type of apraxia, it was found that ideomotor apraxia is the most common type in CBS, whereas ideational apraxia could be present in an advanced stage (Grijalvo-Perez & Litvan, Citation2014). Considering the distinction between conceptual and production systems in praxis functions (Roy, Square, Adams, & Friesen, Citation1985), apraxia in CBS seems to affect the production system. In contrast, no severe impairments of the conceptual system (i.e. semantic memory of gestures) are typically found (Jacobs et al., Citation1999; Salter, Roy, Black, Joshi, & Almeida, Citation2004; Soliveri, Piacentini, & Girotti, Citation2005; Stamenova, Roy, & Black, Citation2011). Concerning gesture modalities, CBS patients usually obtain worse results when tested by imitation rather than by pantomime (Jacobs et al., Citation1999; Peigneux et al., Citation2001; Spatt, Bak, Bozeat, Patterson, & Hodges, Citation2002; Stamenova et al., Citation2011). Moreover, difficulties in gesture execution usually improved when CBS patients could use concrete tools (Graham, Zeman, Young, Patterson, & Hodges, Citation1999; Jacobs et al., Citation1999; Leiguarda et al., Citation2003; Spatt et al., Citation2002; Stamenova et al., Citation2009). Finally, as for the type of gestures, no clear differences in the amount of apraxia between transitive and intransitive gestures (Buxbaum, Kyle, Grossman, & Coslett, Citation2007; Jacobs et al., Citation1999; Leiguarda et al., Citation2003; Peigneux et al., Citation2001) as well as between representational and non-representational gestures (Buxbaum et al., Citation2007; Leiguarda et al., Citation2003; Merians et al., Citation1999; Salter et al., Citation2004; Spatt et al., Citation2002; Stamenova et al., Citation2009) were found.
Visuospatial impairment is another distinctive cognitive feature of CBS (Burrell et al., Citation2014), and it is included in the current diagnostic criteria, i.e. the Mayo Clinic criteria (Boeve et al., Citation2003) and the modified Cambridge criteria (Mathew et al., Citation2011). In particular, CBS patients show difficulties in performing visuospatial tasks, such as Benton’s judgment of line orientation (Soliveri et al., Citation1999) or the spatial tasks of the Visual Objects Spatial Performance (VOSP) battery (Bak, Caine, Hearn, & Hodges, Citation2006). Moreover, constructional apraxia and hemineglect are frequently reported (Boeve et al., Citation2003; Tang-Wai et al., Citation2003). Finally, there are some case reports of CBS patients who showed partial or complete Gerstmann’s syndrome (i.e. digit agnosia, left-right disorientation, agraphia, and acalculia) (Di Stefano et al., Citation2016; Tang-Wai et al., Citation2003) and/or Balint-Holmes syndrome (i.e. oculomotor apraxia, optic ataxia, simultanagnosia, and deficits on estimating depth and distances) (Mendez, Citation2000; Rajagopal et al., Citation2011), which include some visuospatial impairments together with impairment of other abilities (Vallar, Citation2007).
Alien limb syndrome, which is characterized by the sensation that a limb is foreign or has a will of its own and/or by complex unintentional limb movements interfering with normal tasks (Armstrong et al., Citation2013) is another typical disorder in CBS (Armstrong et al., Citation2013; Grijalvo-Perez & Litvan, Citation2014; Murray et al., Citation2007). Several additional phenomena have been described in CBS that are likewise regarded as manifestations of alien hand syndrome (e.g. magnetic apraxia, intermanual conflict, grasping, impulsive hand groping, and purposeless wandering of the limb) (Grijalvo-Perez & Litvan, Citation2014). Interestingly, while CBS patients with frontal dysfunction may show alien limb movements characterized by continuous tactile pursuits of the examiner’s hand (“tactile mitgehen”), the alien limb of patients with more parietal damage is more likely to drift or levitate and assume odd postures (Delrieu et al., Citation2010; Grijalvo-Perez & Litvan, Citation2014; Semenza, Citation2003).
Aphasia is a very common impairment in CBS (Di Stefano et al., Citation2016; Frattali, Grafman, Patronas, Makhlouf, & Litvan, Citation2000; Graham et al., Citation2003; Mahapatra, Edwards, Schott, & Bhatia, Citation2004; McMonagle, Blair, & Kertesz, Citation2006; Murray et al., Citation2007), and typically it is of the non-fluent type (Graham et al., Citation2003; Mahapatra et al., Citation2004; McMonagle et al., Citation2006; Tree & Kay, Citation2008). Moreover, longitudinal studies on aphasia in CBS suggested that patients either show non-fluent speech production from the first presentation (Tree & Kay, Citation2008) or are initially anomic and then develop disorders of expressive language resembling a progressive non-fluent aphasia (McMonagle et al., Citation2006). In contrast, receptive language and single word comprehension are relatively preserved, except in few cases (Di Stefano et al., Citation2016; Graham et al., Citation2003; McMonagle et al., Citation2006).
Regarding the remaining cognitive domains, even though some executive and memory impairments can be found in CBS, many studies have failed to find a specific pattern of prefrontal dysfunction and amnesia. Therefore, the evaluation of these cognitive functions is regarded as being worthless to the differential diagnosis of CBS and other diseases (Burrell et al., Citation2014). Finally, modifications of behavior and personality, similar to those reported in frontotemporal dementia (FTD), are common in CBS (Burrell et al., Citation2014).
Taking into account the brain pathology underlying CBS, it appears with great heterogeneity (Boeve, Citation2011; Boeve et al., Citation2003; Lee et al., Citation2011; Ouchi et al., Citation2014). Specifically, corticobasal degeneration (CBD)—a neurodegenerative condition characterized by atrophy, gliosis, and tau-immunoreactive pathology in the gray and white matter of the neocortex, basal ganglia, and substantia nigra (Murray et al., Citation2007)—is the most frequent underlying pathology. However, other neuropathologies have been reported, including Alzheimer’s disease (AD), frontotemporal lobar degeneration (FTLD), progressive supra-nuclear palsy (PSP), and Creutzfeldt-Jakob disease (CJD) (Boeve, Citation2011; Boeve et al., Citation2003; Lee et al., Citation2011; Ouchi et al., Citation2014).
Bearing in mind the pathological heterogeneity underlying CBS and in hopes that disease-modifying drugs for neurodegenerative dementias will be soon available, detailed clinic-pathological correlation studies are promoted for the purpose of recognizing an early clinical/cognitive marker of a specific pathology. In-depth investigations of CBS clinical and/or cognitive phenotypes are also important for many other reasons. First, few detailed descriptions of CBS have been reported to date. Second, CBS has often been regarded as clinically heterogeneous (Burrell et al., Citation2014; Di Stefano et al., Citation2016), but definite variants of the syndrome have rarely been acknowledged (e.g. symmetric CBS, Dopper et al., Citation2011). Third, many features of the syndrome (e.g. alien hand syndrome, left–right disorientation) have been described exclusively by clinical-observational exams and lack a quantitative validation. Fourth, there are few studies including longitudinal, quantitative assessments of apraxia (Stamenova et al., Citation2009) and other cognitive impairments in CBS (Huang, Hornberger, Hodges, & Burrell, Citation2014). Fifth, an in-depth syndrome description is important because the pattern of cognitive and motor impairments could determine how and to what level the patient fails in performing her/his everyday activities (see, for example, Shakespeare, Yong, Foxe, Hodges, & Crutch, Citation2015 in the realm of Posterior Cortical Atrophy syndrome). In this regard, few data are available of the functional impact of CBS on everyday skills (Stamenova et al., Citation2009). Finally, since the clinical phenotype of dementia is most likely dictated by the topographical distribution of degeneration in the brain (Ouchi et al., Citation2014; Weintraub & Mesulam, Citation2009), an accurate report of the clinical syndrome and its progression over time could identify which core regions are targeted by degeneration and in which directions it spreads along in the brain.
Therefore, the overall aim of this study was to fill the gaps reported above with an in-depth, longitudinal investigation of a clinical and cognitive phenotype of an early onset dementia patient presenting with CBS. The more specific aims of the study were as follows: (i) a detailed evaluation of the cognitive impairments by adopting quantitative tests whenever possible; (ii) an in-depth, longitudinal, and quantitative evaluation of apraxia; (iii) an appraisal of the impact of apraxia and other cognitive impairments on patient functional status; (iv) an indirect mapping of the degeneration spreading in a case of CBS, starting from data on symptom progression as well as clinic-anatomical correlations reported in the literature.
2. Case description
Patient CG came to our attention at approximately 18 months post-disease onset (T1). She was a 53-year-old woman with 8 years of education. Her past medical history was unremarkable. In particular, there was no history of stroke, traumatic brain injury, or alcohol or drug abuse. Moreover, family history was negative for dementia. Only a brief minor depressive episode occurred more than 20 years before. At the first visit, CG reported some difficulties in performing complex actions that all seemed to involve arms movements (e.g. problems in opening the clotheshorse, errors in setting the table, difficulties folding clothes, and slight indecisions in dressing and ironing as well as writing). These reports were confirmed by her relatives, who specified that the symptoms had an insidious onset when CG was approximately 51 years old (approximately 18 months earlier) and showed a progressive course. On the other hand, no fluctuations of cognition and attention were reported. CG was fully aware of her difficulties and consequently very agitated and worried. A neurological examination showed bilateral limb apraxia and upper-limb plastic hypertonia and hyperreflexia. The Mini-Mental State Examination (MMSE) showed a mild cognitive decline (MMSE = 22/30). A neuropsychological evaluation confirmed bilateral ideomotor apraxia and preserved insight (Table ). Moreover, the evaluation showed some deficits in visuospatial abilities and very mild prefrontal executive impairments. Conversely, no clear signs of amnesia, aphasia, visual agnosia and topographical disorientation emerged. Finally, all the aspects of orientation (i.e. temporal, spatial, contextual, and personal) were preserved. A behavioral examination showed mild anxiety and depression but was negative for psychotic symptoms and behavioral disturbances (Neuropsychiatric Inventory, NPI = 10). Functional status was reduced slightly, especially in some activities of daily living (Table ). A brain MRI-scan showed initial focal atrophy of bilateral parietal cortices (Figures and ). An electroencephalogram (EEG) highlighted a diffuse and moderate slowing of the brain activity. A preliminary diagnosis of neurodegenerative dementia not otherwise specified was formulated, and therapies with donepezil, paroxetine and alprazolam were initiated.
Table 1. Longitudinal neuropsychological assessment
Table 2. Evaluation of functional status
Figure 1. CG’s brain imaging. A selection of axial slices from brain MRI scan performed at the first assessment (top) and follow-up visit about one year later (bottom), where focal and increasing bi-parietal cortex atrophy is clearly visible.
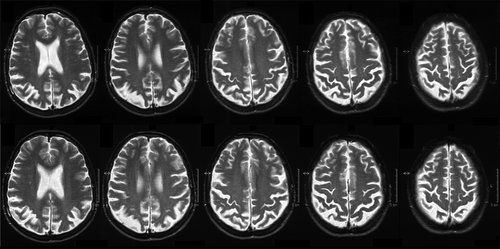
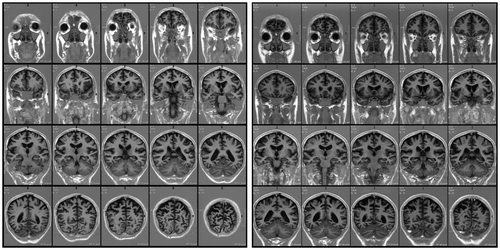
Figure 2. Coronal slices from the first (left) and follow-up (right) MRI examinations further showing posterior bi-parietal atrophy and demonstrating relative sparing of medial temporal and frontal cortices.
A follow-up visit was performed approximately one year later at 32 months post-disease onset (T2). Difficulties in performing complex motor actions were increased (e.g. CG had fallen while catching the bus, had difficulties opening the car door and fastening her seatbelt, had burned her fingers by ironing, had grasped the receiver upside down and wrapped herself in telephone wire), and involuntary, brief muscle contractions of both her arms, closely resembling myoclonus, had been noticed for the first time. Rare odorous misperceptions (e.g. bad smells from fresh-cooked foods made her sick) and simple odorous hallucinations (i.e. unreal bad smells in her home) had also appeared. A neurological examination highlighted extra-pyramidal hypertonia, especially in the left upper limb, hyperreflexia more severe on the left side, deficits in discriminating tactile stimuli applied to symmetrical parts of the body, agraphesthesia, limb apraxia more severe in the left upper limb, and dysdiadochokinesia. The MMSE score was slightly decreased (19/30). A follow-up neuropsychological examination showed that ideomotor apraxia, visuospatial impairments, and prefrontal executive impairments had all worsened (Table ). Moreover, the examination confirmed that apraxia had become asymmetric, with the left arm more affected. Also, mild aphasia emerged for the first time. On the other hand, orientation and insight were still preserved, and no signs of amnesia, visual agnosia, and topographical disorientation emerged. A follow-up behavioral assessment showed that anxiety and depression had improved, but moderate apathy emerged (NPI = 8). Functional status had worsened, especially in the IADL scale (Table ). A control brain MRI scan highlighted mild atrophy to bilateral parietal cortices (Figures and ). FP-CIT (dopamine transporter) SPECT brain imaging showed normal tracer uptake in basal nuclei. A diagnosis of CBS according to all the current diagnostic criteria (Mathew et al., Citation2011), i.e. Toronto criteria (Lang, Riley, & Bergeron, Citation1994), Mayo Clinic criteria (Boeve et al., Citation2003) and modified Cambridge criteria (Bak, Hodges, & Thomas, Citation2008; Mathew et al., Citation2011), was formulated. Finally, levetiracetam was prescribed to treat myoclonus.
A third evaluation was performed approximately one year later, at 49 months post-disease onset (T3). The errors in performing complex motor activities had further increased (e.g. the patient was no longer able to sit in the back seat of her car and showed some difficulties in sitting in the front seat as well; she had difficulties picking up foods with a fork). Moreover, CG’s relatives reported that she sometimes postured with her trunk tilted leftwards, both while on her feet and walking. The brief occurrence of this abnormal posture closely resembled a short-lived episode of trunk dystonia known as Pisa tower syndrome. Besides, CG’s relatives noticed that her left arm sometimes was quite inactive (i.e. a probable sign of motor neglect) and other times stayed somewhat raised (i.e. a probable sign of levitation). Furthermore, they reported that myoclonus of the upper limbs had improved slightly with levetiracetam therapy and that it was more severe in the left limb than in the right limb at this time. Finally, they noticed that odorous misperceptions and simple odorous hallucinations had completely disappeared. A follow-up neurological examination showed hypomimia, fixed gaze, dysarthria, camptocormia, extrapyramidal hypertonia of the four limbs (more severe on the left side), hyperreflexia, at-rest myoclonus of the upper limbs (more severe on the left side), and limb apraxia. The MMSE score was unchanged (21/30). A follow-up neuropsychological assessment showed that many of CG’s cognitive impairments had worsened (Table ). In particular, both ideomotor apraxia and visuospatial impairments reached a severe grade, and aphasia reached a moderate grade. Moreover, insight was slightly impaired for the first time. To the contrary, executive prefrontal impairments were unvaried and still mild, and both visual agnosia and topographical disorientation were still absent. Finally, because of aphasia, it was difficult to test both amnesia and orientation at this time. However, no signs of severe amnesia emerged at the clinical-observational examination (e.g. no perseverations in discourse, no oblivion to recalling autobiographical information). Moreover, orientation was tested by using an ad hoc recognition version of the spatiotemporal orientation test (i.e. a multiple-choice task), instead of the standard free recall version, and CG tested as being unimpaired. A behavioral examination was still negative for behavior disturbances and psychiatric symptoms (NPI = 1). Functional status was severely reduced on the IADL scale (Table ). A brain CT scan confirmed moderate cortical atrophy, especially to bilateral frontal-parietal cortices. A brain HMPAO SPECT scan showed a severely decreased blood flow to bilateral temporal cortices, especially to the right superior temporal gyrus and to bilateral parietal-occipital cortices. A very mild decreased blood flow also emerged in the cuneus, precuneus and bilateral posterior cingulate gyri. Homozygosity of the epsilon-3 allele of the APOE gene resulted from the genetic testing. A globally slow and unstructured brain activity emerged at a follow-up EEG. A levodopa challenge test was negative.
At successive follow-up visits, both motor and cognitive impairments had progressively and rapidly worsened. In particular, three years after the first visit, at approximately 60 months post-disease onset (56 years of age), the MMSE score was 9/30, and a neuropsychological examination was not administrable because of severe dementia. At that time, by GC’s relatives’ wishes, no further examinations were performed.
3. Cognitive assessment
CG underwent the first neuropsychological evaluation at approximately 20 months post-disease onset. Two follow-up evaluations were carried out at 32 and 49 months post-disease onset. A supplementary neuropsychological evaluation was performed to refine the pattern of cognitive impairments at the same time as the second evaluation. Table shows the results of the longitudinal neuropsychological assessment, and Table shows the results of the supplementary neuropsychological examination. CG and her daughter gave their written consent to participate in the study. The study was approved by the Ethical Committee of the Casa di Cura Quarenghi and was done in accord with The Code of Ethics of the World Medical Association (Declaration of Helsinki) for experiments involving humans.
Table 3. Supplementary neuropsychological assessment
3.1. Global cognitive functioning
Global cognitive functioning was assessed using the Mini-Mental State Examination (MMSE) (Folstein, Folstein, & McHugh, Citation1975). The results showed a mild global cognitive decline at the first evaluation that remained relatively unvaried at the successive follow-ups up to the third evaluation at 49 months post-disease onset (Table ). Then, one year later, at approximately 60 months post-disease onset, a more remarkable cognitive decline emerged that reached the stage of severe dementia (MMSE = 9/30). Interestingly, despite the cognitive decline being mild during the first 49 months of the disease course, severe selective neuropsychological syndromes (i.e. apraxia and visuospatial attention disorders) were already present at 18–20 months post-disease onset and subsequently showed a relatively rapid worsening over time.
3.2. Praxis processing
3.2.1. Limb and limb-kinetic apraxia
Limb (ideomotor) and limb-kinetic apraxia were examined employing the De Renzi test (De Renzi, Citation1985; De Renzi, Motti, & Nichelli, Citation1980). It is a test of concurrent or immediately delayed imitation of intransitive gestures that comprises two sections: one involving gestures by arms-and-hands (limb-apraxia) and the second involving gestures by fingers (limb-kinetic apraxia). Moreover, both the presentation and execution gestures are unilateral, e.g. the examiner moves her/his right (or left) arm to probe the movements of the right (or left) arm of the patient. Thus, the De Renzi test returns four partial scores, two of them relating to left and right arm-and-hand movements (limb-apraxia), and the other two scores relating to left and right finger movements (limb-kinetic apraxia). Two total scores, each corresponding to the sum of the partial scores obtained at the two sections of gestures for each body side, are calculated.
A more comprehensive assessment of ideomotor apraxia was accomplished by also administering the task of Pantomimes tool use to verbal command (De Renzi, Faglioni, Scarpa, & Crisi, Citation1986; De Renzi et al., Citation1980). Fifteen tools (e.g. a knife, toothbrush, hammer, comb) were included, and two attempts for the execution of each pantomime were offered to the patient. CG’s performance was scored as 3 points (correct pantomime), 2 points (correct but imprecise pantomime), 1 point (vaguely similar pantomime) or 0 points (wrong pantomime). The total score of each tool was the sum of the scores obtained with the two attempts. The single-item score range was 0–6, and the total test score range was 0–90.
Also, a longitudinal study of limb and limb-kinetic apraxia was carried out by considering CG’s results of three successive administrations of the De Renzi test and the task of Pantomimes tool use to verbal command over a period of three years. A statistical analysis of the longitudinal scores of the De Renzi test and pantomime task was performed by applying Friedman’s ANOVA. Post hoc tests were computed using a Wilcoxon signed-rank test applying the Bonferroni correction.
The results showed that CG’s performance of the De Renzi test of imitation of intransitive gestures was impaired at the first evaluation (Tables and ), supporting an early diagnosis of limb and limb-kinetic apraxia. Then, starting from the second evaluation, a diagnosis of ideomotor apraxia was also confirmed by CG’s impaired performance on the pantomime task (Table ). The longitudinal study showed a significant worsening of limb apraxia as well as limb-kinetic apraxia over time, both considering the results of the imitation test (De Renzi test total score, χ2 = 43.145, df = 5, p < 0.001; De Renzi test limb apraxia score, χ2 = 31.649, df = 5, p < 0.001; De Renzi test limb-kinetic apraxia score, χ2 = 20.837, df = 5, p = 0.001) and the pantomime task (pantomime task, χ2 = 20.844, df = 2, p < 0.001) (Table ).
Table 4. Apraxia longitudinal assessment
Moreover, CG’s total score on the De Renzi test significantly worsened on the second evaluation of the left side of the body (T2–T1 score difference = 53–25, z = −2.751, p = 0.006, two-tailed). No statistical difference was found on the right side (T2–T1 score difference = 53–42, z = −1.580, p = 0.114, two-tailed). Moreover, both left and right limb apraxia significantly worsened on the second evaluation (left arm T2–T1 score difference = 24–1, z = −2.772, p = 0.006, two-tailed; right arm T2–T1 score difference = 27–15, z = −2.058, p = 0.040, two-tailed), but limb-kinetic apraxia remained relatively unchanged (left hand T2–T1 score difference = 29–24, z = −0.850, p = 0.395, two-tailed; right hand T2-T1 score difference = 26–27, z = −0.368, p = 0.713, two-tailed). At the third visit, CG’s total score on the De Renzi test further worsened both for the left and right sides of the body (left side T3–T2 score difference = 25–6, z = −2.565, p = 0.010, two-tailed; right side T3–T2 score difference = 42–24, z = −2.456, p = 0.014, two-tailed). Moreover, limb apraxia results were relatively unchanged (left arm T3–T2 score difference = 1–0, z = −1.000, p = 0.317; right arm T3-T2 score difference = 15–10, z = −1.342, p = 0.180), whereas limb-kinetic apraxia had worsened (left hand T3–T2 score difference = 24–6, z = −2.428, p = 0.015, two-tailed; right hand T3–T2 score difference = 27–14, z = −2.121, p = 0.034, two-tailed).
3.2.2. Other types of apraxia
Oral apraxia was evaluated using a test by De Renzi, Pieczuro, and Vignolo (Citation1966), and gaze apraxia was evaluated through an ad hoc task requiring bi-ocular movements along the horizontal and vertical axes to verbal commands. In detail, four directions of gaze were tested (i.e. upward, downward, rightward and leftward), and CG had to repeat each movement three times. Every single correct movement was scored as one point, and the total range score was 0–12. Regarding other remaining apraxia, truncal, dressing and eyelid apraxia were examined both by observing the patient’s behavior at the visits and by interviewing the caregiver. Moreover, the copy of geometrical figures test (Spinnler & Tognoni, Citation1987) was used to test for constructional apraxia. Finally, ideational apraxia was investigated by means of some items involving multi-step actions taken from the pantomime tool use task as well as the object use task (see below) (De Renzi, Citation1985; De Renzi, Pieczuro, & Vignolo, Citation1968) (e.g. CG had to light a candle with matches, pour a glass of water from a bottle).
Results showed that oral, eyelid, gaze, dressing and truncal apraxia were all absent on the first evaluation. Moreover, ideational apraxia was ruled out because CG had no difficulties on items involving multi-step actions taken from both the pantomime tool use task and the objects use task (see the section Modality of gesture performance: pantomime, imitation and tool use). On the other hand, severe constructional apraxia resulted from the copy of geometrical figure test (Tables and ). On the second evaluation, oral, eyelid and ideational apraxia were still absent, whereas mild gaze dyspraxia emerged (Table ). Moreover, the emergence of dressing apraxia was suggested by CG’s caregiver reports (Table ). Also, some suspicion for truncal apraxia emerged for the first time, especially by noticing some marked hesitations by CG in sitting in the chair in the evaluation room. Finally, constructional apraxia was still severe. On the third evaluation, the total score of the oral praxis test was still in the normal range; nonetheless, some errors emerged for the first time (Table ). Moreover, truncal and dressing apraxia had worsened, and constructional apraxia was of extreme severity. Furthermore, some rare errors in performing multistep actions suggesting a very mild ideational apraxia emerged for the first time with the pantomime tool use task. In contrast, eyelid apraxia was still absent. Finally, gaze praxis was not further evaluated.
3.2.3. Left–right differences
The De Renzi test is a useful tool also for studying left–right differences in limb and limb-kinetic apraxia. In fact, it invites the patient to imitate the same set of intransitive gestures successively with her/his right upper limb and then with her/his left upper limb, and the test accordingly provides separate scores for the performance of the limb of each side of the body. Left–right differences with the De Renzi test were first examined by applying Friedman’s ANOVA. Post hoc tests were computed with the Wilcoxon signed-rank test applying the Bonferroni correction.
Results showed that limb and limb-kinetic apraxia were clearly symmetric on the first examination; in fact, no differences between the left and right sides of the body emerged on the De Renzi test (i.e. left-right total score = 53–53, z = 0.000, p = 1.000, two-tailed; limb-apraxia score = 24–27, z = −0.604, p = 0.546, two-tailed; limb-kinetic apraxia score = 29–26, z = −0.816, p = 0.414, two-tailed) (Table ). However, a significant difference between the right and left body sides, with the left side more impaired, emerged on the De Renzi test total score at the second evaluation (left-right difference = 25–42, z = −2.092, p = 0.036, two-tailed) (Table ). In particular, an asymmetric limb-apraxia emerged with the left upper limb more severely affected than the right limb (left–right difference = 1–15, z = −2.121, p = 0.034, two-tailed), whereas limb-kinetic apraxia was still symmetric (left-right difference = 24–27, z = −828, p = 0.408, two-tailed) (Table ). At the third visit, the left body side was still more impaired than the right side on the total score of the De Renzi test (left-right difference = 6–24, z = −2.058, p = .040) (Table ), but this time both limb apraxia (left-right difference = 0–10) and limb-kinetic apraxia (left–right difference = 6–14) seemed to be asymmetric, even if the amount of the left–right difference did not reach statistical significance both for limb apraxia (z = −1.890, p = 0.059, two-tailed) and limb-kinetic apraxia (z = −1.131, p = 0.258, two-tailed).
3.2.4. Modality of gesture performance: pantomime, imitation and tool use
To advance the study of CG’s apraxia, different modalities of gesture performance were tested (i.e. pantomime, object use, and imitation). The task of pantomimes tool use to verbal command and the parallel task of object use were administered to CG. The first task has been already described in the previous section Limb and limb-kinetic apraxia. The object use task involved 10 concrete tools similar to a subset of those utilized in the pantomime tool use task. In particular, the concrete objects were placed successively on the table in front of CG, and she had to grasp them and show to the examiner how they should be used. The same scoring procedure as the pantomime task was adopted. In particular, two attempts for each object use demonstration were given to the patient. CG’s performance was scored as 3 points (correct demonstration), 2 points (correct but imprecise demonstration), 1 point (vaguely similar demonstration) or 0 points (wrong demonstration). The total score of each tool was the sum of the scores obtained from the two attempts. The single item score range was 0–6, and the total test score range was 0–60.
A comparison between the pantomime and object use tasks was performed on the second visit. A Wilcoxon signed-rank test was applied to test for differences in the patient’s performance.
To compare imitation and pantomime, CG’s ability to pantomime to verbal command 10 meaningful intransitive gestures, taken from the De Renzi test, was examined first (e.g. “please, do the sign of the cross”). Then, we compared CG’s performance on the pantomime of these ten intransitive gestures with her performance on the imitation of the same gestures that had resulted from the standard De Renzi test. Differences were computed as the mean of the Wilcoxon signed-rank test.
Results showed that CG’s performance on the object use task seemed to be slightly better than that on the pantomime tool use task (i.e. 51/60 vs. 46/60) (Table ), but even so, this difference did not reach the statistical significance (z = −1.512, p = 0.131, two-tailed). Similarly, the comparison between pantomime and imitation performed at the time of the second evaluation did not return any statistically significant difference (z = −1.342, p = 0.180, two-tailed) (Table ). However, a clear difference in favor of pantomime emerged at an early stage of disease. In fact, the extended version of the pantomime task (i.e. 15 items) resulted as unimpaired at the first evaluation; conversely, the imitation task (i.e. De Renzi test) resulted as impaired at the same evaluation.
3.2.5. Type of gestures: Transitive vs. intransitive, meaningful vs. non-meaningful
The type of gesture was included as a variable in the evaluation of CG’s apraxia. In particular, a comparison between transitive gestures taken from the test of pantomime tool use to verbal command and intransitive gestures taken from the test of pantomime meaningful gestures to verbal command (see the preceding section) was performed at the second evaluation. A Wilcoxon signed-rank test was used to test for differences.
In addition, since the De Renzi test (De Renzi, Citation1985; De Renzi et al., Citation1980) involves an equal number (i.e. 12) of meaningful (e.g. the sign of victory) and non-meaningful gestures (e.g. put the thumb between the index and middle fingers), a comparison between the two types of gestures was performed. A Wilcoxon signed-rank test was adopted.
The results showed that CG’s performance of transitive gestures on the pantomime tool use task was slightly worse than the performance on the intransitive gestures of the pantomime meaningful gestures task (respectively, 46/60, equivalent to 76.6%, and 28/30, equivalent to 93.3%) (Table ). Moreover, this last difference was statistically significant (z = −2.235, p = 0.025, two-tailed). Regarding the comparison between meaningful and non-meaningful gestures, results did not show any significant differences. In particular, although both types of gestures were slightly impaired on the first examination (Table ), no differences emerged between them when considering both the right and the left upper limbs (right side, z = −0.212, p = 0.832, two-tailed; left side, z = −637, p = 0.524, two-tailed). At the second evaluation, an advantage in the meaningful gestures seemed to emerge exclusively for the right arm (i.e. 27/36 vs. 15/36) (Table ), but this difference did not reach statistical significance (z = −1.000, p = 0.317, two-tailed). At the third evaluation, both types of gestures were severely impaired, but no differences emerged between them (right side, z = −0.137, p = 0.891, two-tailed; left side, z = −1.000, p = 0.317, two-tailed) (Table ).
3.2.6. Input: Recognition of gestures
Recognition of gestures was studied using a task of gesture naming by action at the second evaluation. In particular, the task called for CG to name 10 meaningful intransitive gestures (e.g. military greeting, stop gesture, the sign of the cross) taken from the De Renzi test that were pantomimed by the examiner. In addition, we took into account the results by CG in naming 20 pictures belonging to the categories of tools (e.g. hammer, saw, pliers) as well as musical instruments (e.g. violin, piano, trumpet) (tool naming task) taken from the picture naming test (Laiacona, Barbarotto, Trivelli, & Capitani, Citation1993) at all three successive examinations.
The results showed that CG had no difficulties in recognizing all of the 10 intransitive meaningful gestures taken from the De Renzi test (score = 30/30) at time of the second evaluation. Interestingly, at the same time, CG’s performance on imitation of the same 10 intransitive meaningful gestures on the standard version of the De Renzi test was impaired, especially for the left upper limb (i.e. left upper limb score = 12/30, right upper limb score = 24/30). Regarding the results from the tool naming task, GC’s performance on the 20 items belonging to the categories of tools and musical instruments was relatively unimpaired at both the first (score = 18/20) and the second examinations (score = 17/20). In contrast, mild deficits on the tool naming task suggest a possible initial impairment of the conceptual system of praxis at the third examination (score = 15/20). However, the language impairment found in naming at this time could better explain this result.
3.2.7. Functional impact
The impact of apraxia on CG’s functional status was studied by interviewing the caregiver and using the basic and instrumental activities of daily living scales (ADL, Katz, Downs, Cash, & Grotz, Citation1970; IADL, Lawton & Brody, Citation1969).
The results showed that an early mild decline in CG’s functional status was evident since the first evaluation (Table ). Moreover, CG’s performance resulted as impaired, especially on those activities of her daily life that mainly involved arm movements (e.g. problems in opening the clotheshorse, errors in setting the table, difficulties in folding clothes, slight indecisions in dressing and ironing, difficulty writing) (Table ). This fact suggests that CG’s functional decline was probably strictly related to limb apraxia. Then, at successive visits, a progressive decline in CG’s functional status was documented, probably due to a concurrent worsening of apraxia. However, starting from 32 months post-disease onset, CG’s caregiver frequently reported errors suggesting not only apraxia but also impairment of other cognitive functions, especially visuospatial functions (Table ). Therefore, it is probable that the reduction in CG’s competence in performing activities of her daily life was related not only to apraxia but also to other multiple cognitive impairments starting from an intermediate stage of disease onset.
3.3. Visuospatial processing
3.3.1. Spatial abilities as a whole
Several tests were used to evaluate CG’s visuospatial abilities (Tables and ). In particular, tests of constructional apraxia, visuospatial attention impairments, unilateral neglect, (dorsal) simultanagnosia, and optic ataxia were administered. Moreover, the possible emergence of complex syndromes (i.e. Balint–Holmes’ syndrome and Gerstmann’s syndrome) was checked by using tests of both visuospatial impairments and other different cognitive impairments (e.g. left–right disorientation, acalculia). Finally, data concerning CG’s abilities of topographical orientation were obtained by interviewing the caregiver.
The results showed that visuospatial function disorders were the second more important impairment in CG’s cognitive profile. In fact, several different spatial ability impairments emerged (e.g. constructional impairments, visuospatial attention disorders, unilateral neglect, simultanagnosia). However, it should be noted that some spatial abilities were unimpaired (i.e. hand–eye coordination and topographical orientation).
3.3.2. Constructional functions impairments
A test of drawing geometrical figures was used to test for constructional apraxia (i.e. Copy of geometrical figures test, Spinnler & Tognoni, Citation1987). The results showed severe impairment in the visuoconstructional functions starting from the first evaluation (Table ). Noteworthy, closing-in was one of the most frequent errors. In particular, CG had a strong tendency to draw lines inside the models as if her hand was captured by them (magnetism).
3.3.3. Visuospatial attention disorders
Two tests of visual search were administered to CG for assessing selective attention as well as visuospatial attention (i.e. digit cancellation test, Spinnler & Tognoni, Citation1987; Bell’s test, Gauthier, Dehaut, & Joanette, Citation1989). Moreover, further data about CG’s visuospatial attention impairments were reported by the caregiver and were collected by observing her behavior at the visits.
The results showed that CG’s performance on the two visual search tests was moderately impaired (Table ) at the first examination and then worsened over time. In particular, CG seemed not to be able to efficiently explore the visual space, such that many omissions of targets resulted in the visual search tests. Moreover, a severe difficulty to follow printed stimuli from line-to-line emerged. Furthermore, distractibility was another relevant feature of CG’s attention disorders: in fact, her attention was often captured by task-irrelevant events during the visits. Interestingly, distractibility was not associated with a more general environmental dependency; in fact, utilization and imitation behaviors (Iaccarino, Chieffi, & Iavarone, Citation2014; Lhermitte, Citation1983) never appeared. Another aspect of CG’s attention disorders was a difficulty in disengaging her attention from stimuli (sticky fixation) (e.g. when she has to cross a street, she stares at a point at the far side of the street for a long time before starting to move). Finally, a pathological narrowing of the visuoattentional focus (tubular vision) also seemed to emerge.
3.3.4. Unilateral neglect
Unilateral neglect was assessed through a visual search test (i.e. Bell’s test, Gauthier et al., Citation1989) and by considering possible position-preference effects on the Raven progressive colored matrices test (Basso, Capitani, & Laiacona, Citation1987). In detail, “position-preference effect” refers to the tendency of a patient with left (or right) unilateral neglect to pick drawings especially on the right (or left) side of a page on the Raven test. To further evaluate unilateral neglect, CG’s performance on writing and reading tasks used in the assessing of aphasia was considered. Conversely, CG’s performance in drawing was not usable because of severe constructional apraxia.
Regarding the results, a mild left unilateral neglect emerged on Raven’s test that showed a significant position-preference effect on right-sided items at the first evaluation (Table ). Then, at the second evaluation, left visuospatial neglect was confirmed, with both a more pronounced position-preference effect with right-sided items on the Raven test and the occurrence of some errors with neglect point on a reading task (AAT battery). At the third evaluation, Bell’s test was positive for left unilateral neglect for the first time, and further errors with neglect point emerged on the reading task. Moreover, signs of motor neglect exclusively affecting the left arm were reported by the caregiver and confirmed on the observational examination (e.g. CG’s left arm assumed a quite passive posture and remained inactive during the visit).
3.3.5. Simultanagnosia
Simultanagnosia was evaluated using an object decision task (i.e. chimerical figures) taken from the Birmingham Object Recognition Battery (BORB, Riddoch & Humphreys, Citation1993). Moreover, the dot counting task taken from the Visual Object and Space Perception Battery (VOSP, Warrington & James, Citation1991) was administered to examine the spatial abilities further.
Results showed that a mild impairment emerged with the object decision task (Table ) as well as with the Dot counting task at the second evaluation (Table ). Such results were suggestive of simultanagnosia, especially considering that CG’s performances on concurrent tasks of visual processing, both at a pre-associative (i.e. discrimination of Scribble test) and associative stage (i.e. picture-naming task) (see the section Visual-perceptual processing), were all unimpaired. Moreover, since CG suffered from marked visuospatial impairments more than visual perception impairments and had never read letter-by-letter, as is usually done by ventral simultagnosics (Riddoch et al., Citation2010), we hypothesized that her simultanagnosia was of the dorsal type.
3.3.6. Optic ataxia
A simple reaching task was devised and administered to CG for assessing optic ataxia. In particular, CG had to reach and grasp an object (e.g. a little bottle of water) with her hand that was placed alternatively at one of three different locations (i.e. in high, middle, and low positions) in both her left and right visual hemifields. Moreover, CG had to execute the test under both the conditions of central (foveal) and peripheral vision and alternatively by using her right arm and her left arm. One point was scored for any appropriate grasping. The total score for each hand was 12 points (6 locations × 2 conditions). The results showed that CG’s performance on the reaching task was fully unimpaired (Table ). Thus, optic ataxia was ruled out.
3.3.7. Balint–Holmes’ syndrome
A complete Balint–Holmes’ syndrome (see Vallar, Citation2007) never appeared; nonetheless, some features of the syndrome emerged. In particular, some signs of gaze apraxia and a suspected narrowing of the visuospatial attentional focus was noticed in the observational examination at the second visit. Moreover, simultanagnosia resulted in the object decision task being taken from BORB. Nonetheless, both optic ataxia and a defective ability in estimating distances and in-depth perception never emerged.
3.3.8. Gerstmann’s syndrome
A complete Gerstmann syndrome (for a review see Rusconi, Pinel, Dehaene, & Kleinschmidt, Citation2010) emerged starting at the second evaluation. The emergence of the four impairments of the syndrome (i.e. finger agnosia, left–right disorientation, dysgraphia, and acalculia) was supported by the results of quantitative testing (see the sections Finger agnosia, left–right disorientation, Written language processing, and Number processing).
3.4. Body awareness processing
3.4.1. Finger agnosia
Finger gnosis was tested using a modified version of the task for finger localization by Spinnler and Tognoni (Citation1987). In the standard version of the task, the patient’s hand is covered, and the examiner touches simultaneously two of her/his out-of-sight fingers. The patient’s task is to point to the fingers on a drawing of her/his hand that she/he feels touched by the examiner. However, since CG showed severe difficulties in pointing, probably because of apraxia, we had to modify the standard procedure of the task and train CG to name the out-of-sight fingers that she felt was touched by the examiner instead of pointing them on a drawing of the hand as in the standard version.
Results showed that CG’s performance was severely impaired in the modified version of the finger localization task, especially, but not exclusively, for the left hand (Table ). These results were suggestive for finger agnosia. An alternative explanation based on CG’s difficulty in naming related to aphasia seemed less convincing. In fact, CG suffered from mild aphasia at that time, whereas her performance on the finger gnosis task was severely impaired. Moreover, omissions and other signs of word-finding problems (e.g. circumlocutions, verbal and semantic paraphasias, semantic conduite d’approche) were rare (i.e. 2 omissions out of 12 items). Finally, CG was able to retrieve the names of no less than four different fingers (i.e. the thumb, forefinger, middle finger, and little finger) without any word-finding hesitations during the visit.
3.4.2. Autotopagnosia
Body schema abilities were examined using a shortened version of Semenza and Goodglass (Citation1985) battery, which included some tests of naming as well as of pointing to body parts. More details are that two tasks of naming body parts (respectively on patient’s body and on examiner’s body) and two tasks of pointing to body parts (respectively, on patient’s body and on examiner’s body) were administered (Table ). Both the tasks of naming and pointing to body parts on the patient’s body had to be executed with eyes open in the first section and with eyes closed in the second section. A crossed-pointing task was also administered in which CG had to use her right hand to point to some parts on her left hemi-body and her left hand to point to some parts on her right hemi-body (Table ).
Results showed that both tasks of naming body parts were relatively unimpaired (Table ). Also, the three pointing tasks of the Bisiach battery (i.e. the two tasks of pointing to body parts and the crossed-pointing task) all resulted in being relatively unimpaired (Table ). Therefore, a diagnosis of autotopagnosia was excluded. Moreover, no differences emerged between the conditions of open and closed eyes in the task of pointing to body parts on the patient’s body.
3.4.3. Left–right disorientation
Left–right disorientation was assessed using the same tasks utilized for assessing autotopagnosia. The pointing tasks included in the Bisiach battery called for the patient to point to alternating right or left body parts. In the same way, the patient had to declare the parts of the left vs. right side of the body pointed to by the examiner in the naming tasks of the battery. Therefore, the results from these tests were also used to identify possible left–right disorientation. Moreover, the crossed-pointing task from the same battery was also an appropriate tool for the assessing of left–right discrimination.
The results showed that CG had severe left–right disorientation. In fact, the tasks from the Bisiach battery requiring left–right discrimination were all impaired (Table ). On the whole, CG made 30 errors out of the 48 left–right discriminations requested (62.5%).
3.4.4. Agonistic dyspraxia
Awareness and representation of body parts were studied only in a preliminary way overall by observing CG’s behaviors and reactions while she was performing tests involving limbs and hands movements, especially the De Renzi test of limb and limb-kinetic apraxia (De Renzi, Citation1985; De Renzi et al., Citation1980).
The results were somewhat unexpected. In fact, a phenomenon similar, but not coincident, to what other authors previously described and named as agonistic dyspraxia (Aboitiz et al., Citation2003) emerged. In particular, when a request of executing a movement of a forelimb (for example, the right one) is made, the patient suffering from agonistic dyspraxia correctly executes that movement but does it by using her/his contralateral forelimb (the left one in the example) instead of the correct forelimb that remains immobilized. Moreover, this contralateral movement is experienced as involuntary by the patient, who has serious difficulties in controlling it. Finally, agonistic dyspraxia occurs only when unilateral movements are requested by a verbal command and not under imitation or spontaneous execution. Similar to the descriptions of agonistic dyspraxia, when CG had to perform a movement with her left or right hand, the other hand (the right or the left, respectively) replaced it and automatically or compulsively executed that movement, performing the same purposeful action in its place (i.e. the agonistic hand). CG had difficulties counteracting these substitutions, and after she corrected herself, a new replacement occurred. As a consequence, her behavior appeared as being hesitant. This phenomenon of replacing was evident both under imitation and verbal command and only occasionally presented also in spontaneous movement. However, in a quite different way from patients with agonistic dyspraxia (Aboitiz et al., Citation2003), the substitutions managed by the agonistic arm and hand of CG seemed to start exclusively after that the correct arm had begun its movement, just like in mirror movement (see Boeve et al., Citation2003). It seemed that the agonistic arm of CG was called to action in a mirroring way, but as soon as it started to imitate what the other arm was doing, this last arm stopped moving. Therefore, simultaneous concurrent movements of both arms, as it occurs in the classical mirror phenomenon (see Boeve et al., Citation2003), never appeared. Interestingly, there emerged no differences in agonistic dyspraxia between the left and right arm and hand up to the second visit (i.e. the left hand replaced the right one and vice versa with the same frequency). In contrast, at a more advanced stage of disease, CG’s right hand replaced the left one more often.
3.4.5. Other phenomena
The possible occurrences of intermanual conflict, as well as of the anarchic and alien hand syndromes, were studied by interviewing CG’s caregiver and by observing her spontaneous behavior at the visits and during her performance on the De Renzi test.
The results showed that, even if CG experienced involuntary movements like those described above (i.e. agonistic dyspraxia), an experience of estrangement from and personification of the limbs never appeared (i.e. alien hand). Moreover, CG sometimes experienced hand and arm movements as being slightly unwilled but never as being strange and uncooperative (i.e. anarchic hand). Furthermore, no feelings of having additional fingers or limbs emerged (i.e. supernumerary hand). In addition, bimanual coordination was preserved, and CG had no problems performing actions requiring simultaneous cooperation of both hands (e.g. opening a bottle of water, closing her pencil case, putting on and off a pen’s cap, cutting a sheet of paper with a scissors, opening a padlock with a key). Additionally, we never observed one of CG’s hands performing actions to the contrary and in an opposite way to (i.e. diagonistic dyspraxia), or interfering with (i.e. intermanual conflict), the actions performed by the other hand. Also, although clear signs of somatic personal neglect (i.e. hemisomatoagnosia) never appeared, some signs of motor neglect affecting the left upper limb were noticeable at the observational examination starting at the second evaluation. Furthermore, at the time of the second evaluation, CG’s caregiver reported that CG’s left forearm remained frequently raised to approximately 45 degrees when she was holding her bag (i.e. levitating hand). Finally, magnetic errors (i.e. magnetic apraxia) frequently emerged on the De Renzi test, without differences between the left and right body side, at all the three evaluations, and a more general magnetism was confirmed in CG from many occurrences of the closing-in phenomenon when drawing (see Ambron & Della Sala, Citation2017; Conson, Salzano, Manzo, Grossi, & Trojano, Citation2009). On the whole, all the symptoms collected (i.e. bilateral agonistic dyspraxia, left arm levitation and bilateral magnetic apraxia) resembled a mild corticobasal or posterior variant of alien hand syndrome (Semenza, Citation2003).
3.5. Language processing
3.5.1. Oral language processing
The language was examined using the picture-naming test (Laiacona et al., Citation1993) and by the Achener Aphasia Test (Luzzatti, Willmes, & De Bleser, Citation1996).
Results showed overall that aphasia was a salient aspect of the CG’s cognitive profile, even if less prominent and early when compared to impairments of other cognitive domains. In fact, aphasia started only from the second evaluation and was never severe. Specifically, no signs of aphasia appeared in the conversational speech during the first evaluation; only rare hesitations of articulation and infrequent and brief word-finding latencies were noticeable. Auditory comprehension was preserved. No difficulties emerged with the repetition item of the MMSE, and the picture naming test was unimpaired (Table ).
At the second visit, speech was fluent with normal utterance length, but its rate was slowed by word-finding difficulties. Moreover, frequent use of imprecise words was evident, and negligible impairments of articulation, as well as minimal signs of paragrammatism, were also noticeable. Furthermore, some suspects for auditory comprehension impairment emerged with conversation. Regarding the results of the AAT, repetition was globally preserved with some minor difficulties exclusively emerging with the longest sentences. Moreover, no significant deficits emerged in the naming task (Table ). In contrast, moderate impairments of both auditory and written comprehension were confirmed in the Token test as well as in the comprehension task of the AAT. On the whole, a profile resuming transcortical sensory aphasia of mild gravity resulted from the data collected.
At the third evaluation, aphasia had worsened, and its profile had changed to mixed transcortical aphasia of moderate gravity. The conversational speech appeared globally fluent; nonetheless, brief, stereotyped and often interrupted utterances, together with mild articulatory problems, were evident. Moreover, mild echolalia and more clear signs of paragrammatism, as well as some phonological paraphasias, were present. Furthermore, both word-finding difficulties and auditory comprehension impairments had worsened in conversation. Regarding the results of testing, the deficits in both the picture naming test (Laiacona et al., Citation1993) and the naming task of the AAT had worsened (Table ). Moreover, the impairment in the Token test as well as in the comprehension tests of the AAT became severe. Finally, repetition was still minimally impaired, with some slight difficulties emerging exclusively with the longest sentences.
3.5.2. Written language processing
Written language processing was assessed only in a preliminary way. In particular, three tasks of written language included in the AAT battery were administered: a task of reading of words (both regular and irregular) as well as of brief sentences, a task of writing with tokens (the tokens representing single letters in a first section of the task and words in a second section), and finally a task of handwriting to dictate. Moreover, spontaneous handwriting was also examined.
No difficulties emerged on the reading of simple sentences, like those included on the MMSE, at the first evaluation. At the second evaluation, the reading abilities resulted as minimally impaired, especially in the task of reading brief sentences on the AAT (score = 26/30). Moreover, some errors containing the neglect point (Ellis, Flude, & Young, Citation1987) emerged, suggesting very mild neglect dyslexia. At the third evaluation, the performance on the reading task of the AAT was more clearly impaired (score = 17/30). In particular, there were many errors, especially in the reading of sentences as well as of irregular words (i.e. reading irregular words scored 1/3 compared to a score of 3/3 in reading regular words). Moreover, errors in neglect point were also frequent. Consequently, a diagnosis of peripheral dyslexia, probably neglect dyslexia, was confirmed, and a preliminary diagnosis of surface dyslexia was suggested for the first time by considering the selective errors in the reading of irregular words. Unfortunately, the reading task included a limited number of items (i.e. only 3 regular and 3 irregular words) and did not include non-words; therefore, a more accurate diagnosis of dyslexia cannot be made.
Regarding the results of writing abilities, CG’s performance resulted as being severely impaired (score = 3/30) in the task of writing words and brief phrases under dictation, starting from the second evaluation. In contrast, only a minimal impairment in the task of writing with tokens under dictation (score = 24/30) emerged at the same evaluation. Concomitantly, severe difficulties emerged in the spontaneous handwriting of single words, and the handwriting of sentences was unfeasible. At the third evaluation, CG’s dysgraphia had notably worsened, and any further examination was not possible. Taking into account a qualitative analysis of CG’s errors in writing, as a whole, CG’s handwriting was almost completely illegible. Specifically, most of her errors involved letter morphology and suggested allographic impairments (e.g. the use of upper- and lower case letters in the same word). Moreover, frequent scrawls emerged in CG’s writing because either the letters in a word often overlapped and/or the lines of a single letter were perseverated (i.e. motor, continuous perseverations). Furthermore, spatial errors characterized especially by misalignment also emerged. In contrast, micrography was absent. In sum, taking into account the characteristics of CG’s spontaneous handwriting, as well as the dissociation that resulted between the two dictation tasks, with a severe impairment on handwriting and a relative preservation in writing with tokens, a diagnosis of peripheral dysgraphia (i.e. apraxic agraphia) was formulated (Passov, Gavrilova, Strand, Cerhan, & Josephs, Citation2011). Unfortunately, because of the severity of peripheral distortions, a study of possible forms of central dysgraphia was unfeasible.
3.5.3. Number processing
CG’s arithmetical abilities were examined using a brief test battery for acalculia included in the Esame Neuropsicologico per l’Afasia (Capasso & Miceli, Citation2001). It involves some different tasks of number processing (i.e. repetition, reading, dictating and transcoding tasks) as well as some common arithmetical operations (i.e. addition, subtraction and multiplication).
Results were strongly indicative of acalculia: among the seven tasks included in the ENPA battery, only the task of repetition resulted in being unimpaired (Table ). In particular, a diagnosis of acalculia, including impairment of both number-processing and calculation, was formulated.
Regarding number-processing, both number production (i.e. writing of numbers from dictation) and comprehension (i.e. reading of numbers) were impaired. Moreover, selective impairment of graphemic processing, both of Arabic numerals and verbal numerals, seemed to emerge. Furthermore, a qualitative analysis of the errors showed that both syntactic and lexical errors were evident, both in the reading and writing of numbers. Besides, severe difficulties emerged also in the transcoding task. In contrast, no impairment of phonological processing emerged (i.e. repetition was preserved). Regarding impairments of calculation, they seemed to involve the retrieval of both arithmetical facts and calculation procedures. In contrast, no disorder in processing arithmetical symbols emerged.
3.5.4. Pre-frontal executive processing
Executive pre-frontal functions were evaluated by means of a task of working memory (digit span backward; Orsini et al., Citation1987), the phonological fluency test (Novelli et al., Citation1986), a verbal semantic fluency task (Spinnler & Tognoni, Citation1987) and the Cognitive Estimate Test (CET; Della Sala, MacPherson, Phillips, Sacco, & Spinnler, Citation2003). Moreover, logical deductive visual reasoning was investigated using Raven’s colored progressive matrices test (Basso et al., Citation1987).
The results showed that some pre-frontal functions were slightly impaired, starting at the second evaluation. However, executive impairments were very mild, and a complete dysexecutive syndrome never appeared; therefore, these impairments should be considered as a secondary aspect of CG’s cognitive profile. In particular, a selective mild deficit of working memory resulted in digit span backward at the first evaluation. Conversely, the other tests of executive functioning were unimpaired (Table ). Moreover, CG preserved good insight. Finally, the results of Raven’s colored progressive matrices test were at all times disputable because of the emergence of a preference effect for right-side items, probably due to unilateral neglect.
At the second evaluation, executive impairments were slightly more evident. In particular, some deficits emerged both in the working memory task and the cognitive estimates test. Noteworthy is that CG’s performance on the verbal fluency tasks was still in the normal range; moreover, she still preserved full insight. At the third evaluation, executive impairments had worsened slightly (Table ); moreover, CG showed partial insight for the first time.
As described above, CG’s pre-frontal function impairments involved the executive functions exclusively, and apart from a mild bradyphrenia, compelling signs of akinetic and disinhibited pre-frontal syndromes never appeared.
3.5.5. Memory processing and orientation
Anterograde long-term memory was assessed using the prose recall test (Spinnler & Tognoni, Citation1987), and verbal short-term memory was assessed using the forward digit span test (Orsini et al., Citation1987). Results showed that long-term memory was one of the best preserved cognitive domains. In fact, the anterograde verbal memory test (prose recall) resulted as unimpaired. Moreover, memory problems were never reported by CG’s caregiver, and perseverations in the discourse or other signs of episodic amnesia never emerged from the observational examinations at the successive visits. Furthermore, signs of retrograde amnesia never emerged by interviewing CG. Also, the unimpaired performance on the picture naming test supported the preservation of semantic memory. However, a slight impairment in short-term memory emerged on the digit span test at the third visit (Table ). Regarding orientation, it emerged that personal, familial, temporal, spatial and contextual orientations were all preserved at the first and second evaluations. At the third visit, it was difficult to test orientation because of aphasia. Nevertheless, orientation was tested by recognition instead of free recall, and CG did not make any errors.
3.5.6. Visual-perceptual processing
Visual perception was studied using the discrimination of Scribbles test (Spinnler & Tognoni, Citation1987) and by analyzing CG’s object recognition performance on the picture-naming task (Laiacona et al., Citation1993).
Results showed that visual perception was one of the best preserved cognitive domains. The caregiver never reported difficulties of CG at recognizing and identifying familiar faces as well as known objects or places. Moreover, the task of apperceptive agnosia (Scribble discrimination test, Table ) resulted in the normal range at the time of the second evaluation, supporting the preservation of an early stage of visual processing. Furthermore, CG’s performance on object recognition on the picture naming task was unimpaired; thus, the emergence of associative agnosia was ruled out (Table ). A mild deficit in the picture naming task emerged at the third visit; nevertheless, the errors were attributable to aphasia and not to agnosia.
4. Discussion
4.1. The cognitive phenotype of CBS
4.1.1. General
This single case study supports the notion that cognitive impairments are a very relevant feature of CBS. In fact, the longitudinal neuropsychological assessment showed a significant cognitive dysfunction, with some cognitive impairments starting at an early stage of disease. Moreover, the complex pattern of cognitive impairments demonstrated by CG overlapped the pattern usually described in CBS (Graham et al., Citation2003; Murray et al., Citation2007). In particular, CG’s cognitive profile was dominated by limb apraxia, which was associated with limb-kinetic as well as constructional apraxia from an early stage of disease. Moreover, gaze, truncal and dressing apraxia were present starting from a later stage of disease. In contrast, oral, eyelid, ideational and conceptual apraxia were relatively absent. Visuospatial skills disorder, especially involving visuospatial attention impairments, constructional apraxia, mild unilateral neglect and simultanagnosia, was the second most important impairment of CG’s cognitive profile. In contrast, topographical disorientation never appeared. Aphasia was another salient impairment, even if it was not a prominent aspect at an early stage. It first was diagnosed with mild transcortical sensory aphasia and then changed into mixed transcortical aphasia of moderate severity. Moreover, early and severe agraphia, probably of the peripheral type, together with very mild neglect dyslexia, was associated with aphasia. Another relevant syndrome of the CG’s cognitive phenotype was a posterior variant of alien hand syndrome (Semenza, Citation2003). It comprised different phenomena, including (left) levitating hand, magnetic apraxia and an impairment similar to what some authors named agonistic dyspraxia (Lavados et al., Citation2002). Furthermore, a complete Gerstmann syndrome emerged, characterized by acalculia, finger agnosia without autotopagnosia, left–right disorientation and agraphia, and a partial Balint’s syndrome, including gaze dyspraxia, simultanagnosia and a narrowing of the visual attention focus but without manifest optic ataxia and no apparent deficits in estimating distances and in depth perception. Considering the secondary aspects of CG’s cognitive phenotype, some executive impairments emerged, starting at the second visit. However, insight was preserved, and compelling signs of the akinetic or disinhibited pre-frontal syndromes never appeared. The remaining cognitive domains appeared well preserved. In particular, signs of amnesia (i.e. anterograde, retrograde and semantic amnesia), disorientation (i.e. spatial, temporal, personal, familial and contextual disorientation), and visual agnosia (both apperceptive and associative) never emerged.
4.1.2. Apraxia
To the best of our knowledge, there are few studies in which limb apraxia has been assessed longitudinally using a standardized test in CBS patients as made in this study (Stamenova et al., Citation2009). In doing so, not only the relevance and severity of limb apraxia in CBS was confirmed by precise quantitative data, but also its pejorative and relatively rapid course over time was adequately traced. Moreover, quantitative measures of apraxia allowed us to make accurate comparisons between the left and the right upper limb performance. Thus, robust data about the issue of symmetry vs. asymmetry of apraxia along the disease course were obtained (see Section 5).
Regarding the results about limb-kinetic apraxia in CG’s case, it should be noted that since the De Renzi test includes items also investigating finger movements, it was considered an adequate tool for evaluating not only limb but also limb-kinetic apraxia (Borroni et al., Citation2008a; Soliveri et al., Citation2005). In this respect, data from this study confirmed the relevance of limb-kinetic apraxia in CBS and its association with limb apraxia (Borroni et al., Citation2008a; FitzGerald et al., Citation2007; Leiguarda et al., Citation2003; Soliveri et al., Citation2003, 2005). Moreover, CG’s case highlighted that limb-kinetic apraxia could have an early onset in CBS and that it could also be bilateral instead of unilateral as usually reported (Stamenova et al., Citation2009). Furthermore, the different drop in time traced between limb and limb-kinetic apraxia supported the distinction between these two forms of apraxia as well as the notion of limb-kinetic apraxia as a separate clinical entity.
As for other apraxias in CG’s case, on the whole, this study suggested a relative heterogeneity. First, apraxia also affected movements of body parts different from the arms (limb-apraxia) and the fingers (limb-kinetic apraxia). For example, clear signs of gaze and truncal apraxia emerged in CG’s case, as similarly reported in other CBS cases (Okuda et al., Citation2001; Rajagopal et al., Citation2011). Second, apraxia seemed to strongly contribute to an impairment of complex functions, such as handwriting (apraxic agraphia), drawing (constructional apraxia) and dressing (dressing apraxia) in CG’s case. However, relative preservation of orofacial praxis was confirmed (Stamenova et al., Citation2009). Regarding the analysis of type as well as mode of gestures, CG’s case study seems to confirm previous data showing that imitation was more affected than pantomime in CBS (Jacobs et al., Citation1999; Peigneux et al., Citation2001; Spatt et al., Citation2002; Stamenova et al., Citation2011). In fact, even though the comparison between pantomime and imitation tasks was not statistically significant at the time of the second evaluation, a clear difference emerged between them early on at the first evaluation, when the pantomime task resulted as unimpaired and the imitation task was clearly impaired and suggestive of limb apraxia (De Renzi test). The fact that imitation was exclusively impaired constitutes an original finding in a CBS patient, considering that usually both pantomime and imitation are reported as impaired in CBS (Stamenova et al., Citation2009). Moreover, this finding suggests that an imitation task could be the most sensitive to detect limb apraxia in an early stage of CBS. Taking into account the comparison between pantomime and object use, no statistically significant difference emerged; nonetheless, a small advantage of object use seemed to emerge, in agreement with many previous data (Graham et al., Citation1999; Jacobs et al., Citation1999; Leiguarda et al., Citation2003; Spatt et al., Citation2002; Stamenova et al., Citation2009). Regarding the comparison between transitive and intransitive gestures (with pantomime), it emerged that both types of gestures were affected, but transitive gestures were more affected, as already found in previous CBS cases (Chainay & Humphreys, Citation2003; Pharr et al., Citation2001; Salter et al., Citation2004). As to the distinction between meaningful (representational) and meaningless (non-representational) gestures, the results showed that both types of gestures were equally affected, just as reported in other CBS patients (Buxbaum et al., Citation2007; Leiguarda et al., Citation2003; Merians et al., Citation1999; Salter et al., Citation2004; Spatt et al., Citation2002; Stamenova et al., Citation2009).
Regarding the possible accounts of apraxia, some further conclusions seemed to emerge from the CG’s case study. First, following the apraxia information-processing model proposed by Roy (Citation1996), CG’s case study further confirms the view that apraxia in CBS was characterized by an impairment in the production system overall in an early and/or intermediate stage of disease with a concurrent preservation of the conceptual system (Stamenova et al., Citation2009). Forthcoming damage to the conceptual system seemed to emerge in a late stage of disease in CG’s case, as signaled by the errors in the tool naming task. Moreover, the study of the different task modalities and gesture types shows that overall CBS affects the direct route to imitation at an early stage of disease with relative preservation of the indirect route. CG concurrently showed, in fact, impaired imitation of meaningless gestures (direct route) and relative preservation of pantomimes (indirect route). The concurrent impairment of the imitation of meaningful gestures found in CG’s case could be explained, as suggested by other authors (Stamenova et al., Citation2011), either from the patient being in some way forced to use the direct route to imitation or from a disconnection between hypothetical input and output semantic systems (input and output praxicon) in gesture processing (Gonzalez Rothi, Ochipa, & Heilman, Citation1991). Finally, a definite impairment of the pantomimes, also signaling disruption of the indirect route of gesture production, was evident starting at an intermediate stage of the disease.
4.1.3. Visuospatial functions impairment
CG’s case study confirms the relevance of visuospatial skill disorders in CBS; in fact, CG showed many significant visuospatial impairments, including visuoconstructional impairments, visuospatial attention disorders, unilateral neglect, and probably simultanagnosia. Considering that visual agnosia and severe dyslexia were absent, CG’s case supports the view that CBS is a “where” stream disorder that is against the preservation of the “what” stream (Bak et al., Citation2006; Possin, Citation2010). Moreover, CG’s case draws attention to visual-spatial attention impairments, a feature less frequently reported in CBS. Actually, some previous descriptions of CBS patients included features that could suggest visual spatial attention impairments, such as being unable to follow printed material from line-to-line (Tang-Wai et al., Citation2003), correctly cancel a target in a field of letters (FitzGerald et al., Citation2007) or find objects within visual space (Rajagopal et al., Citation2011). However, CG had striking and primary visuospatial attention disorders characterized by an inefficient exploration of the visual space, distractibility, problems in disengaging attention (i.e. sticky fixation) and a suspected narrowing of the visual attentional focus. These elements strongly suggest a dysfunction of the posterior attentional system (PAS, Posner & Petersen, Citation1990), the complex system comprising cortical and sub-cortical centers (i.e. posterior parietal cortex, especially lateral parietal cortex, thalamic nuclei connected with the pulvinar and the reticular nucleus, and superior colliculus) that manage the spatial components of visual attention.
4.1.4. Posterior alien hand syndrome
What the mild form of alien hand syndrome showed by CG is known as a corticobasal or posterior variant of alien hand syndrome (Kloesel, Czarnecki, Muir, & Scott Keller, Citation2010; Semenza, Citation2003), and it has already been reported in other CBS cases (Delrieu et al., Citation2010). The posterior alien hand syndrome of CG included left arm levitation, magnetic apraxia (Denny-Brown, Citation1958) and an impairment similar to a condition that some authors have named agonistic dyspraxia (Aboitiz et al., Citation2003). However, magnetic apraxia, which is a well-known form of the alien limb phenomenon (FitzGerald et al., Citation2007), was very mild in CG’s case and only consisted of some involuntary reaching behaviors with her hands in some tests and some occurrences of the closing-in phenomenon in the drawing. Agonistic dyspraxia in CG consisted in “substitutions of movements” for which, as soon as one of her forelimbs and/or hands (left or right) started to perform a requested movement (invariably on verbal command, imitation, and sometimes also in spontaneous execution), the other limb (right or left, respectively), probably after being activated in a mirrored way, replaced it and automatically or compulsively performed the same action in its place.
Regarding other manifestations of alien hand syndrome, CG never exhibited phenomena pertaining to frontal magnetic apraxia, like a compulsive tactile exploration of objects, grasping and groping behaviors, and imitation and utilization behaviors (Lhermitte, Citation1983; Lhermitte, Pillon, & Serdaru, Citation1986). Moreover, other features of the classical alien hand syndrome (e.g. alien and anarchic hand, intermanual conflict) were absent. Finally, the emergence of a posterior variant of alien hand syndrome in CG’s case was consistent with her primary bi-parietal neuropsychological dysfunction and the mild impairment of more anterior functions, like pre-frontal executive functions.
4.1.5. Aphasia
As reported in the introduction section, aphasia is a very common impairment in CBS (Frattali et al., Citation2000; Graham et al., Citation2003; Mahapatra et al., Citation2004; McMonagle et al., Citation2006; Murray et al., Citation2007) and is typically of the non-fluent type (Graham et al., Citation2003; Mahapatra et al., Citation2004; McMonagle et al., Citation2006; Tree & Kay, Citation2008). Despite this apparent general agreement about the pattern of language impairments in CBS, a relative heterogeneity emerged among aphasic syndrome diagnoses in the studies in which an accurate classification of aphasia was possible. Actually, all the main classic aphasic syndromes seemed to be reported, in at least some CBS cases (i.e. global, Broca, transcortical motor, conduction, anomic, Wernicke, and transcortical sensory aphasias) (Frattali et al., Citation2000; Graham et al., Citation2003; Kimura et al., Citation2008; McMonagle et al., Citation2006). CG’s case seems to confirm this heterogeneity of the aphasic presentations in CBS. In fact, although the transcortical sensory aphasia shown by CG at an early stage of disease has already been reported in some previous CBS cases (Graham et al., Citation2003; McMonagle et al., Citation2006), to the best of our knowledge, there are no preceding reports of mixed transcortical aphasia in CBS at an intermediate or a more advanced stage. Specifically, aphasia in CG’s case was fluent, with prominent word-finding difficulties and comprehension deficits. Nonetheless, it showed a progressive conversion from a posterior (e.g. anomic aphasia) to a more anterior aphasia, with some features of non-fluent aphasia added at an intermediate stage (e.g. brief, stereotyped and often interrupted utterances; mild articulatory problems; signs for paragrammatism), similar to what is recorded in the course of aphasia in other CBS cases (McMonagle et al., Citation2006).
Therefore, CG’s case report supports the view that aphasic syndrome in CBS could strictly depend on the time of evaluation, and there might be a tendency to find the non-fluent type at an intermediate or a more advanced stage of disease. Vice versa, the fluent type could be present at an earlier stage of the disease, as shown in CG’s case. In conclusion, caution is required when assessing a typical aphasic syndrome in CBS, irrespective of the evaluation time along the disease course. Interestingly, according to Catani et al.’s hypothesis (Catani, Citation2005; Catani, Jones, & Ffytche, Citation2005), mixed transcortical aphasia could be provoked by damage to the posterior segment of the indirect pathway of the arcuate fasciculus linking Wernicke’s area to the inferior parietal lobule and concurrently to the anterior segment (in CG’s case probably partially) linking the inferior parietal lobule to Broca’s area. This model aligns with the possible origin of mixed transcortical aphasia in CG. In fact, damage to left inferior parietal lobule in CG’s case probably extended to deep white matter and thus possibly intercepted the posterior segment of the indirect pathway of the arcuate fasciculus.
4.2. CBS clinical heterogeneity: A posterior variant
4.2.1. Diagnostic criteria for CBS
CG exhibited a complex syndrome characterized by an insidious onset and a progressive course that involved motor (i.e. rigidity, myoclonus, and truncal dystonia), cortical motor-sensory (i.e. apraxia, number processing deficits, cortical sensory loss, and posterior alien hand syndrome), and cognitive features (i.e. language impairment, visuospatial deficits and mild pre-frontal dysfunction) that are levodopa-resistant and associated with early focal parietal and later parietal-frontal atrophy on brain imaging. Moreover, albeit initially symmetric, the motor (i.e. rigidity and myoclonus) and cortical motor-sensory features (i.e. limb apraxia and posterior alien hand syndrome) became asymmetric, with the left side more affected than right one, starting at the second evaluation at 32 months post-disease onset. From this stage on, CG’s clinical picture assumed an asymmetrical course. The whole clinical phenotype described at 32 months post-onset fully resembles a diagnosis of CBS according to all the current accepted diagnostic criteria (Mathew et al., Citation2011) (i.e. Toronto criteria (Lang et al., Citation1994), Mayo Clinic criteria (Boeve et al., Citation2003) and modified Cambridge criteria (Bak et al., Citation2008; Mathew et al., Citation2011)). Moreover, CG’s syndrome resembles the clinical phenotype of possible corticobasal syndrome recently outlined by some authors (Armstrong et al., Citation2013) in their proposal of new criteria for the diagnosis of CBD.
4.2.2. Symmetry vs. asymmetry
Interestingly, CG could be viewed as an unusual case of CBS because her motor symptoms had a bilateral and symmetric onset. In fact, parkinsonism and myoclonus affected both her arms equally at CBS presentation. Also, limb and limb-kinetic apraxia (Table ), as well as the mild posterior alien hand syndrome, were first bilateral, affecting both upper limbs with the same gravity. In full agreement, a strongly symmetrical parietal atrophy appeared on neuroimaging. The CBS-defining feature of marked asymmetry of the motor symptoms (Mahapatra et al., Citation2004), together with asymmetry of limb apraxia (Table ) as well as of posterior alien hand syndrome, emerged only at the second visit, approximately 30 months post-disease onset. Then, CBS assumed the usual progressive asymmetric course with both motor and motor sensory-cortical features affecting the left limb more than the right one. At this time, an initial mild asymmetric hypometabolism (right hemisphere > left hemisphere) was found on the brain SPECT scan. The symmetrical onset as well as the asymmetrical course of disease in this study was supported by quantitative data from the De Renzi test of apraxia that did not show any left–right difference at the first evaluation and did show more severe apraxia to the left limb at all the subsequent assessments (Table ). Interestingly, Dopper et al. (Citation2011) described the case of a 61-year-old male patient with an atypical CBS presentation, including symmetrical rigidity, ideational and ideomotor apraxia and later dystonia in his left leg, without alien hand phenomenon and myoclonus. This clinical presentation paralleled mild symmetrical cerebral atrophy, especially frontoparietal and perisylvian, on MRI and symmetrical frontoparietal hypometabolism on SPECT. Moreover, the patient was found to be a sporadic case of FTLD-TDP type 3 pathology due to a novel progranulin mutation. The authors named this case as symmetrical CBS. CG’s case study replicates the finding by these authors (Dopper et al., Citation2011) of a symmetrical CBS (S-CBS), even when considering the first part of the disease course. Moreover, this replication extends this finding to a more classic CBS. A patient studied by (Dopper et al., Citation2011) in fact exhibited some unusual features in respect to a classical CBS (e.g. marked behavioral changes, memory impairment, and preserved visuospatial functions) but lacked other typical CBS signs and symptoms (e.g. myoclonus and alien hand syndrome). Another finding from CG’s case study suggests that cases of S-CBS might not be so rare and atypical if patients were studied at a very early stage of disease. In fact, the time interval between disease onset and the first visit in both CG’s case and the case described by Dopper and co-authors (i.e. respectively 18 and 24 months post-disease onset) was relatively briefer than those in other CBS case series (e.g. 32 months post-disease onset, Murray et al., Citation2007; 36 months post-disease onset, Borroni et al., Citation2008b; 48 months post-disease onset, Stamenova et al., Citation2009; 84 months post-disease onset, Stamenova et al., Citation2011). Therefore, in agreement with the symptoms progression in CG’s case, which was first symmetric and became asymmetric at approximately 30 months post-onset, it cannot be excluded that typical asymmetric CBS reported at a middle stage of disease course, when patients are usually studied, actually would have been preceded by an atypical symmetric CBS presentation. This hypothesis seems to be plausible for those patients whose motor and motor-sensory cortical symptoms were bilateral but more severe on a defined body-side at a middle disease stage (relative asymmetry). In contrast, it obviously appears to be less plausible for those patients who presented with focal symptoms at a middle disease stage (absolute asymmetry).
Regarding another previous study that is relevant to the issue of symmetry/asymmetry, some authors retrospectively selected five patients with symmetrical clinical presentations from an autopsy-proven CBD case series and compared them with other classical asymmetric CBD patients of the same series (Hassan et al., Citation2010). All symmetric CBD cases had symmetric brain atrophy on MRI scans and an absence of asymmetric frontoparietal hypometabolism and/or hypoperfusion on PET and/or SPECT scans. Age of onset of symmetric CBD was more praecox (i.e. mean age of 61 years, with a range of 40–69 years vs. a mean age of 66 years with a range of 61–73 years); moreover, it had a similar duration but a more frequent family history of dementia. Phenotypes of symmetric CBD cases were: atypical AD (one case), bvFTD (three cases) and FTDP (one case). Behavioral changes were prominent in symmetric CBD cases and core features of CBS, such as myoclonus, dystonia, limb apraxia and alien limb phenomenon, were all absent. Based on these findings, the authors suggested that symmetric CBD (S-CBD) may be different from the more typical asymmetric variant presenting as CBS.
No pathological data were available in CG’s case, and this was a limitation of the study. However, irrespective of the brain pathology, this study demonstrates that the attribute of symmetry can be associated with an otherwise classical CBS over a reliable period of the disease course, and more generally, that symmetric and asymmetric phenotypes can be associated with the same syndrome/disease at different moments of its course. Thus, CG’s case study suggests that caution should be used in using symmetry or asymmetry as a defining feature of CBS and/or CBD. In other words, the evidence of an S-CBS suggests not stressing asymmetry in the diagnostic criteria of CBS overall at onset or at an early stage of the disease, which are the most important times for diagnosing dementia. On the other hand, this study confirms the association between an asymmetric course and CBS starting at an intermediate stage of the disease.
4.2.3. CBS and PCA
CBS and PCA are two distinct clinical entities, each one with its current diagnostic criteria (for PCA see Crutch et al., Citation2017). Nonetheless, as previously reported by some authors (Alladi et al., Citation2007), there is an overlap of cognitive features between CBS and PCA, with both syndromes being characterized by apraxia and prominent visuospatial features (e.g. visual neglect, Gerstmann’s syndrome). Moreover, the possible involvement of parietal cortex asymmetry in both clinical variants makes it difficult to clinically distinguish CBS from PCA in certain cases (Alladi et al., Citation2007). Furthermore, clinically diagnosed CBS patients have been reported who show marked visuospatial dysfunction (Bak et al., Citation2006), including signs of Balint’s syndrome (Mendez, Citation2000; Rajagopal et al., Citation2011), which is considered one of the core features of the PCA syndrome (Crutch et al., Citation2017). In addition, at least three pathologically diagnosed CBD patients and one patient without pathologic diagnosis have been reported with a PCA syndrome as a presenting symptom followed by signs of CBS (i.e. asymmetric apraxia and parkinsonism) later in the course of the illness (two patients studied by Tang-Wai et al., Citation2003; one patient by Lee et al., Citation2011; one patient by Giorelli, Losignore, Bagnoli, Difazio, & Zimatore, Citation2014). Finally, a subgroup of PCA patients has been found to show motor features commonly associated with CBS (i.e. limb rigidity and apraxia, myoclonus, tremor and alien limb phenomenon) (Ryan et al., Citation2014).
In this regard, CG’s case study further confirms that there is a close overlap between CBS and PCA syndrome. In fact, CG’s clinical picture comprised many features in agreement with the diagnostic criteria of CBS; nonetheless, it also included some of the core diagnostic features of PCA. Moreover, CG showed many cognitive features of a PCA variant known as the bi-parietal syndrome (i.e. apraxia, visuospatial impairments, agraphia, preserved basic perceptual abilities, and preserved object recognition and reading) (Alladi et al., Citation2007; Crutch et al., Citation2017). As recently noted by some authors (Crutch et al., Citation2017), further studies are necessary to better disentangle PCA syndrome from related syndromes, such as CBS. CG’s case study highlights that a detailed study of the bi-parietal syndrome may play an important role to this aim because it may pertain to both PCA syndrome and CBS.
4.2.4. Posterior variant of CBS (P-CBS)
In sum, CBS in CG’s case showed some atypical features characterized overall by a clear symmetrical presentation and an early posterior global aspect of cognitive and motor cortical impairments (e.g. initial fluent aphasia, posterior alien hand syndrome), included many features of the PCA bi-parietal syndrome, and lacked more anterior CBS-typical features (e.g. alien hand, early non-fluent aphasia, behavioral disturbances, personality changes). Moreover, basal ganglia were apparently not involved, which was found in previous CBS cases (Ceravolo et al., Citation2013; Cilia et al., Citation2011; Hammesfahr et al., Citation2016; Homma et al., Citation2014). Thus, CG’s case study further supports the clinical heterogeneity of CBS’s presentation. Moreover, it suggests the emergence of a specific clinical variant of CBS with pronounced posterior involvement (posterior-CBS), as recently found by some authors who proposed to label it as a Gerstmann variant of CBS (Di Stefano et al., Citation2016). Considering the underlying brain pathology, despite the absence of an e4 allele from the apolipoprotein E (APOE) genotyping, Alzheimer’s disease could be the main disease in CG’s case, as it is frequently found in many previously described CBS patients (from 23 to 50%, Hu et al., Citation2009), In fact, CG showed many of the clinical features suggestive of AD pathology vs. CBD pathology (e.g. young age of onset, occurrence of myoclonus, absence of tremors, severe visuospatial deficits, lack of orofacial apraxia, presence of dysgraphia, and important functional decline) (Burrell, Hornberger, Villemagne, Rowe, & Hodges, Citation2013; Hu et al., Citation2009). Moreover, some studies found that widespread atrophy extending to involve the posterior parietal and temporal lobes, as found in CG’s case, may indicate an underlying AD pathology (Burrell et al., Citation2013; Burrell et al., Citation2014; Di Stefano et al., Citation2016; Josephs et al., Citation2010; Lee et al., Citation2011; McMillan et al., Citation2016; Whitwell et al., Citation2010).
4.2.5. Functional impact of CBS
Few studies have examined the impact of limb and limb-kinetic apraxia on competence and autonomy in performing activities of daily living in CBS patients (Stamenova et al., Citation2009). To this purpose, successive semi-structured interviews, as well as basic (and instrumental) Activities of Daily Living scales (ADL and iADL), were administered to the caregiver. On the whole, the results showed that CBS was associated with a significant reduction of competence in performing activities of daily living at a very early stage of disease (i.e. before 18 months post-disease onset) in CG’s case. Regarding the question of what cognitive impairments were responsible for the functional reduction, some authors underlined that it is difficult to distinguish the impact of apraxia from that of other motor and cognitive impairments that probably contribute to disrupting the performance on ADL in brain-damaged patients (Sunderland & Shinner, Citation2007). With regards to this point, the analysis of CG’s errors in performing activities of daily living strongly suggests impairment of praxis abilities rather than of other cognitive functions, at least for up to 32 months post disease onset (Table ). Moreover, starting from 32 months post onset, CG’s caregiver frequently reported errors highly suggestive of a motor and/or other cognitive impairments and especially of visuospatial functions impairments (Table ). Thus, it is probable that the reduction of CG’s autonomy in her daily life recorded from an intermediate stage and onward would be related not only to apraxia but also to other multiple cognitive impairments. In particular, we believe that the visual-spatial attention impairments played the main role in limiting CG’s functional ability. In fact, they probably forced CG to perform actions in a warped visual spatial attention space and/or upon the wrong objects, toward which her attention focus was automatically engaged and from which she could not disengage her attention. As a consequence, visual spatial attention impairments probably neutralized the advantages offered by affordances to guide action and invite the use of objects (Norman, Citation1988). Therefore, a concurrent disruption of both top-down (praxis) and bottom-up (affordance and invitation to use) processes, which are guides for action, inevitably caused important failures in performing activities of daily living in this CBS case. However, other different explanations cannot be excluded. For example, some authors proposed an interesting account based on the “technical reasoning hypothesis” that postulates that the ability to use tools and objects in daily life depends not only on semantic knowledge about tool function and context of use but also on technical reasoning of mechanical properties of tools and objects (Baumard et al., Citation2016; Goldenberg & Spatt, Citation2009). In particular, they suggested and preliminarily verified that the difficulties in tool use showed by CBS patients can be due to technical reasoning deficits following parietal lobe damage (Baumard et al., Citation2016).
4.3. From symptoms progression to degeneration spreading
4.3.1. Neuroanatomical substrate of CG’s cognitive impairments
The in-depth study of CG’s cognitive impairments let us recognize some specific symptoms and syndromes that were well-known in the clinical literature. At this point, starting from data about known anatomo-clinical correlations, we tried to retrace the map of CG brain regions involved as neuro-anatomical substrates of her symptoms and syndromes (Figure ). Moreover, taking into account the data from the longitudinal cognitive assessment over three years, a brain map of the possible spreading of neurodegeneration in this case of CBS was reconstructed (Figure ). We are aware both that the method used in this section was quite unconventional and that data showed (Figure included) were highly speculative. Indeed, no advanced imaging study was performed to support our conclusions, and this is a limitation of this section of the study.
Figure 3. Longitudinal mapping of degeneration spreading in patient CG. Colored areas represent the brain loci of degeneration that were inferred from CG’s cognitive phenotype and by considering well-known brain anatomical-functional correlations found in the literature. The reported stages refer to the successive evaluations performed in the study.
Regarding the results of the first evaluation, at approximately 18–20 months post-disease onset, the core cognitive impairments were bilateral limb and limb-kinetic apraxia and visuospatial disorders, including visuoconstructional and visuospatial attention deficits. Minimal working memory deficits, suspected elements of an incoming Gerstmann syndrome (writing difficulties and dyscalculia noticed by GC’s relatives) and a suspected mild left visuospatial neglect, were all associated with the core impairments at this time. Therefore, following data about anatomo-clinical correlations, early and preferential damage to the bilateral dorsolateral parietal cortices (Goldenberg, Citation2009), including both the superior and the inferior parietal lobules (probably the more anterior parts) as well as to the bilateral premotor cortices, could be hypothesized. In particular, the left inferior parietal lobule, especially the supramarginal gyrus (BA 40) (ideomotor apraxia) (Clark, Boutros, & Mendez, Citation2005, chapter 4) and the right superior parietal lobule (BA 5 and 7) (visuoconstructional impairment and visuospatial attention impairment) (Clark et al., Citation2005, chapter 4), were surely involved. In contrast, more posterior parts of the parietal cortex (i.e. the left angular gyrus (BA 39)) (Gerstmann syndrome, Clark et al., Citation2005, chapter 4) and the more inferior parts of the inferior parietal lobule (i.e. the right temporoparietal junction) (visuospatial neglect, Bisiach & Vallar, Citation2000) were only minimally involved at the time of the first assessment. Moreover, apart from an early preferential damage targeting the bilateral frontal premotor cortices (BA 6) (limb-kinetic apraxia, Binkofski & Reetz, Citation2008), and a probable involvement also of the frontal eyes fields (BA 8) (visual search deficits, Malloy, Cohen, & Jenkins, Citation1998), the prefrontal dorsolateral cortex (probably BA 8, 9) (working memory impairment) (Clark et al., Citation2005, chapter 6) was only minimally damaged. The analysis of the neural substrate of CBS in CG’s case was supported enough by morphological brain imaging (MRI) data at the time of the first evaluation, which showed mild focal atrophy in bilateral parietal cortices (Figures and ).
At the second visit, 32 months post-disease onset, all core (i.e. limb apraxia and visuospatial impairments) and associated mild or incoming deficits (i.e. dysexecutive syndrome, unilateral neglect, and Gerstmann’s syndrome with dysgraphia, acalculia, left–right disorientation, and digital agnosia) emerged before they had worsened except for limb-apraxia, which appeared as unvaried. Moreover, novel cognitive impairments emerged for the first time: agraphesthesia, truncal apraxia, dressing apraxia, some signs of the Balint’s syndrome (i.e. gaze dyspraxia, simultanagnosia, and narrowing of the field of spatial attention, with the exclusion of optic ataxia), some elements of the posterior alien hand syndrome (i.e. agonistic dyspraxia and magnetic apraxia), and a mild transcortical sensory aphasia. Finally, limb but not limb-kinetic apraxia started to be asymmetric, with the left arm more impaired than the right one. Therefore, regarding the neural substrate, we could hypothesize that damage to the earliest foci of neurodegeneration in dorsolateral parietal cortex (e.g. right superior parietal lobule for dressing apraxia and agraphesthesia) (Clark et al., Citation2005, chapter 4) had increased and that some regions in the right hemisphere were slightly more affected than their left counterparts at the time of the second evaluation. Moreover, the posterior and inferior parts of the parietal cortices, which were partially affected at the first evaluation, i.e. the left angular gyrus (BA 39, Gerstmann syndrome) (Clark et al., Citation2005, chapter 4) and the right temporoparietal junction (visuospatial neglect, Bisiach & Vallar, Citation2000), were probably more diffusely damaged at this time. Furthermore, the amount of neurodegeneration of the bilateral frontal premotor cortices (BA 6) (limb-kinetic apraxia, Binkofski & Reetz, Citation2008) seemed to be equivalent, while the damage to the prefrontal dorsolateral cortex (probably BA 8, 9) (working memory impairment and executive functions) (Clark et al., Citation2005; chapter 6) was slightly increased. Finally, it seemed that degeneration had spread from the earliest core foci on dorsolateral parietal cortices to more posterior and inferior parietal regions, following a direction toward the adjacent occipital and temporal lobes. In fact, multiple sites of damage to the right parietal-occipital cortex (posterior form of alien hand syndrome with agonistic dyspraxia) (Semenza, Citation2003), bilateral parietal-occipital cortices (partial Balint’s syndrome, Clark et al., Citation2005, chapter 4), posterior temporoparietal junction (mild truncal apraxia, Okuda et al., Citation2001) and finally to the left temporal-parietal–occipital carrefour (mild transcortical sensory aphasia, Catani et al., Citation2005) could be hypothesized at the time of the second evaluation. Unfortunately, follow-up brain imaging data were not available at this stage of the disease course.
At the third evaluation, 49 months post-disease onset, limb and limb-kinetic apraxia, as well as visuospatial disorders and prefrontal functions impairments, had all worsened. Moreover, limb apraxia continued to be asymmetric, and limb-kinetic apraxia started to be asymmetric for the first time, with the left side of the body side more affected than the right, just as had occurred before for limb apraxia. Furthermore, other relevant changes emerged at this time: aphasia worsened, and its profile converted into mixed transcortical aphasia of moderate gravity, impairment of verbal short-term memory started to be associated with aphasia, and minimal signs of ideational apraxia appeared for the first time. In addition, the posterior alien hand syndrome worsened, and signs of left levitating hand were also evident at that time. Finally, clear signs of motor neglect affecting the left arm emerged, and a minimal hesitation without significant apraxia resulted in the orofacial praxis test. Regarding the neural substrate underlying these further changes in the clinical phenotype, we first could hypothesize increased damage to the brain regions already targeted by the degeneration. Moreover, our analysis seemed to suggest that degeneration had spread to posterior and inferior regions of the parietal cortex near to the occipital lobe, such as the right parietal-occipital cortex (i.e. more complete posterior alien hand syndrome including left levitating hand, Delrieu et al., Citation2010) and to the temporal lobe, such as left temporoparietal regions (ideational apraxia, Binkofski & Reetz, Citation2008; and short-term memory impairment, Lheman & Schnider, Citation2008). Furthermore, according to Catani et al.’s hypothesis (Catani, Citation2005; Catani et al., Citation2005), mixed transcortical aphasia could be provoked by damage to the posterior segment of the indirect pathway of the arcuate fasciculus linking Wernicke’s area to the inferior parietal lobule and to the anterior segment linking the inferior parietal lobule to Broca’s area. Also, damage to anterior regions, such as premotor cortex (more severe limb-kinetic apraxia, Binkofski & Reetz, Citation2008) or dorsolateral prefrontal cortex (worsening of executive function impairments) (Clark et al., Citation2005, chapter 6), had probably increased at the third evaluation. Finally, the probable symmetry in the amount of degeneration affecting posterior regions at the second evaluation, which caused a more severe left limb apraxia, extended forward to right frontal deep regions (left motor neglect, Goldenberg, Citation2003) and to the right premotor cortex (asymmetric limb-kinetic apraxia, Binkofski & Reetz, Citation2008) at the third evaluation. Interestingly, the severe hypoperfusion in the bilateral temporal cortices, especially the right superior temporal gyrus and in the bilateral parietal-occipital cortices, emerged on SPECT scan at the third evaluation, supporting the hypothesis of a degeneration spreading to more posterior and inferior parietal regions along directions toward the occipital and the temporal lobe, respectively.
Moreover, the results seemed to confirm that the brain damage had become asymmetric, with some regions of the right brain hemisphere more affected than their left counterparts. Finally, bilateral frontoparietal atrophy emerged on the follow-up CT scan, compared to the previous focal bilateral parietal atrophy that had emerged on the MRI scan, supporting that degeneration spreads from posterior parietal regions to anterior frontal regions.
4.3.2. Large-scale brain network hypothesis in CBS
The performed analysis suggests that the bilateral dorsolateral parietal and predominantly lateral frontal cortices, as well as the bilateral prefrontal cortices, are the possible underlying neuroanatomic substrates of the clinical syndrome of CG. Moreover, longitudinal data seemed to suggest that degeneration had started in hubs of the parietal and frontal cortices and then spread toward adjacent regions in the brain. However, a long-distance spreading of degeneration from parietal to frontal (and prefrontal) regions cannot be excluded. In fact, a gradient of disease propagation from posterior (i.e. parietal cortex) to more anterior regions (i.e. premotor cortex) was suggested by the fact that limb apraxia started to worsen and become asymmetric, with the left side more impaired than the right one, about one year before limb-kinetic apraxia. Similarly, the modification of aphasia over time from transcortical sensory aphasia to mixed transcortical aphasia seemed to suggest a gradient of disease propagation from posterior to more anterior regions in the left hemisphere. Therefore, the anatomical substrates underlying the disease propagation in CG’s case were probably not only the short association fibers (i.e. arcuate or U-fibers) connecting neighboring gyri inside the parietal lobe or between the parietal gyri and the adjacent occipital and temporal gyri but also the long association fibers connecting the parietal lobules with frontal and prefrontal cortex, including the frontal eye field (i.e. the superior longitudinal fasciculus) (Bartolomeo, Thiebaut de Schotten, & Chica, Citation2012; Catani, Citation2005; Catani et al., Citation2005; Clark et al., Citation2005, chapter 4; de Schotten et al., Citation2011) as well as the indirect route of the arcuate fasciculus (Catani, Citation2005; Catani et al., Citation2005).
Interestingly, CG’s case study seems to support the hypothesis that neurodegenerative diseases selectively target distinct large-scale intrinsic brain networks (Seeley, Crawford, Zhou, Miller, & Greicius, Citation2009). In fact, the multiple brain regions assumed to be the neuroanatomic substrates in CG’s case (i.e. bilateral dorsolateral parietal and predominantly lateral frontal cortices as well as bilateral prefrontal cortices) matched well with the large-scale intrinsic network involving the dorsal parietal and predominantly lateral prefrontal cortices (i.e. dorsal sensorimotor association network) (De Luca, Beckmann, De Stefano, Matthews, & Smith, Citation2006; Seeley et al., Citation2009) already identified as the best candidate for CBS. Moreover, the longitudinal study seems to suggest that degeneration remained confined to this large primary network for over three years without involving other large-scale networks (e.g. default mode network (Buckner et al., Citation2005; Seeley et al., Citation2009), salience network (Seeley et al., Citation2007)) usually implicated in other diseases (Alzheimer’s disease, AD, and behavioral variant frontotemporal dementia, bvFTD). A proof of this fact is that the precuneus, which is considered one core region in the AD default network, was unimpaired at an early stage of disease and/or only minimally impaired at an advanced stage (see the results from SPECT imaging), despite the fact that it was confined to a core and an early focus of degeneration in CG’s case (i.e. the dorsolateral region of the superior parietal lobule (BA 5 and 7)). Moreover, the fact that degeneration seemed to start and spread inside a unique large-scale functional network gives some clues of a disease propagation mode along trans-synaptic connections, mimicking a prion-protein domino effect in which just (or mainly) neurons functionally connected with dysfunctional neurons become impaired (for a review, see Guo & Lee, Citation2014).
Considering a higher level of detail, the large-scale parieto-frontal network found as the underlying functional network of CBS includes or is associated with more local and specific networks. In particular, a dorsal attentional network (DAN) as well as a ventral attentional network (VAN) (Bartolomeo et al., Citation2012; Corbetta & Shulman, Citation2002; de Schotten et al., Citation2011), an executive-control network (Seeley et al., Citation2007), a medial frontal-prefrontal network (Wolpe et al., Citation2014) and a basal ganglia network (Rittman, Ghosh, & Rowe, Citation2013; Südmeyer et al., Citation2012) have been documented. In this respect, bearing in mind CG’s cognitive phenotype, we think that some of these sub-networks were surely involved in CG’s case, especially a dorsal attentional network (DAN) as well as a ventral attentional network (VAN) (Bartolomeo et al., Citation2012; Corbetta & Shulman, Citation2002; de Schotten et al., Citation2011) and an executive-control network (Seeley et al., Citation2007). However, another local network (i.e. a medial frontal-prefrontal network Wolpe et al., Citation2014) was probably not involved. Moreover, although bilateral extra-pyramidal symptoms were present from the first evaluation, FP-CIT SPECT imaging of CG showed normal tracer uptake in basal nuclei. Thus, data suggested that the basal ganglia network (Rittman et al., Citation2013; Südmeyer et al., Citation2012) was not involved in CG’s case. This finding supports the current hypothesis that suggests extrapyramidal motor symptoms in CBS are not invariably associated with SNc neuronal degeneration and that supra-nigral factors may play a major role in several cases (Ceravolo et al., Citation2013; Cilia et al., Citation2011; Homma et al., Citation2014).
5. Conclusions
CG’s case study confirms that CBS is a heterogeneous syndrome and suggests that a less common variant could emerge (i.e. a posterior CBS (P-CBS)). This somewhat atypical CBS phenotype would be characterized by a symmetrical presentation, an asymmetrical course, and prominent posterior (bi-parietal) cognitive and cortical motor manifestations, including many features of the PCA bi-parietal syndrome and lacking more anterior CBS typical features. Moreover, it seems to lack basal ganglia involvement. Thus, this study demonstrates that the attribute of symmetry can be associated with an otherwise classical CBS for a reliable period of its course and more generally that the attributes of symmetry and asymmetry can be associated with the same syndrome/disease in different moments of its course. Furthermore, the evidence of early (temporary) symmetrical manifestations in the CBS phenotype suggests not stressing “asymmetry” in CBS diagnostic criteria at an early stage of disease, as current clinical criteria do for diagnosing dementia.
The present study offers a further detailed outline of the cognitive phenotype in CBS and its progression over time. Many findings of limb apraxia in CBS have been replicated and substantiated, and some novel findings have emerged, such as distinct trajectories of worsening between the limb and limb-kinetic apraxia, relevant visuospatial attention disorders, the emergence of a posterior alien hand syndrome and mixed transcortical aphasia.
Data collected seem to suggest that apraxia and visuospatial attention disorders have an early and relevant functional impact on the patient performance of activities in daily life. In particular, we made the hypothesis that visuospatial attention impairments could neutralize the advantages of guide for action offered by affordances and invitations to use the objects. Therefore, a concurrent disruption of both top-down (i.e. praxis) and bottom-up (i.e. ineffective affordances and invitations to use the objects due to visuospatial attention impairment) processes that guide the action inevitably caused an early and severe decline in the functional status of CG.
Finally, the present study seems to support the hypothesis that neurodegenerative diseases selectively target distinct large-scale intrinsic brain networks. In particular, clinical data were compatible with an overall dysfunction of the underlying large-scale intrinsic network (i.e. dorsal sensory-motor association network) already identified as a proper candidate of CBS. Moreover, longitudinal data suggested that degeneration might remain confined within this network without involving other neighboring networks and that it spread not only along short association fibers connecting neighboring gyri inside the parietal lobe or between parietal and adjacent occipital and temporal gyri but also through long association fibers connecting the parietal lobules with the frontal and prefrontal cortex, including the frontal eye field (i.e. superior longitudinal fasciculus) and the indirect route of the fasciculus arcuatus.
Funding
The authors received no direct funding for this research.
Acknowledgments
Special thanks are due both to patient CG and her daughter for their longstanding cooperation with interviews, examinations and testing. Moreover, many thanks are due to Luigi Ghilardini for editing digital Figures and and to Silvia Casarotto for her help in editing Figure .
Additional information
Notes on contributors
Carlo Abbate
Carlo Abbate and Pietro Davide Trimarchi are neuropsychologists and clinical researchers, who specialize in dementia diagnosis. Carlo Abbate has a PhD in Physiopathology of Aging at the University of Milan and works at the Fondazione Ca’ Granda, Ospedale Maggiore Policlinico in Milan. Pietro Davide Trimarchi has a PhD in Experimental Psychology and Cognitive Neuroscience at the University of Milano–Bicocca and works at Fondazione Don Carlo Gnocchi, in Milan. They have been working together for years in many research areas: early cognitive markers of dementia, Mild Cognitive Impairment, clinical syndromes of dementia, qualitative neuropsychological examination, and variants of Alzheimer’s disease. The firm belief that neuropsychology can be a helpful tool both to understand and to diagnose dementia has guided their work. The detailed longitudinal study of the cognitive and behavioral status of a young adult patient with degenerative dementia presented here well exemplifies the method adopted by the authors.
References
- Aboitiz, F., Carrasco, X., Schröter, C., Zaidel, D., Zaidel, E., & Lavados, M. (2003). The alien hand syndrome: Classification of forms reported and discussion of a new condition. Neurological Sciences, 24, 252–257. doi:10.1007/s10072-003-0149-4
- Alladi, S., Xuereb, J., Bak, T., Nestor, P., Knibb, J., Patterson, K., & Hodges, J. R. (2007). Focal cortical presentations of Alzheimer’s disease. Brain, 130(10), 2636–2645. doi:10.1093/brain/awm213
- Ambron, E., & Della Sala, S. (2017). A critical review of closing-in. Neuropsychology, 31(1), 105. doi:10.1037/neu0000295
- Armstrong, M., Litvan, I., Lang, A. E., Bak, T. H., Bhatia, K. P., Borroni, B., … Weiner, W. J. (2013). Criteria for the diagnosis of corticobasal degeneration. Neurology, 80, 496–503. doi:10.1212/WNL.0b013e31827f0fd103
- Bak, T. H., Caine, D., Hearn, V. C., & Hodges, J. R. (2006). Visuospatial functions in atypical parkinsonian syndromes. Journal of Neurology, Neurosurgery, and Psychiatry, 77, 454–456. doi:10.1136/jnnp.2005.068239
- Bak, T. H., Hodges, J. R., & Thomas, H. B. (2008). Corticobasal degeneration: Clinical aspects. In C. Duyckaerts & I. Litvan (Eds.), Handbook of clinical neurology (pp. 509–521). Amsterdam: Elsevier.
- Bartolomeo, P., Thiebaut de Schotten, M., & Chica, A. B. (2012, May 4). Brain networks of visuospatial attention and their disruption in visual neglect. Frontiers in Human Neuroscience. doi: 10.3389/fnhum.2012.00110.
- Basso, A., Capitani, E., & Laiacona, M. (1987). Raven’s coloured progressive matrices: Normative values on 305 adult normal controls. Functional Neurology, 2(2), 189–194.
- Baumard, J., Lesourd, M., Jarry, C., Merck, C., Etcharry-Bouyx, F., Chauviré, V., … Le Gall, D. (2016). Tool use disorders in neurodegenerative disease: Roles of semantic memory and technical reasoning. Cortex, 82, 119–132. doi:10.1016/j.cortex.2016.06.007
- Binkofski, F., & Reetz, K. (2008). Apraxia. In S. F. Cappa (Ed.), Cognitive neurology: A clinical textbook (pp. 67–88). USA: Oxford University Press.10.1093/acprof:oso/9780198569275.001.0001
- Bisiach, E., & Vallar, G. (2000). Unilateral neglect in humans. In F. Boller, J. Grafman, & G. Rizzolatti (Eds.), Handbook of neuropsychology (2nd ed., Vol. 1, Chapter 16, pp 459–502). Amsterdam: Elsevier Science B.V.
- Boeve, B. F. (2011). The multiple phenotypes of corticobasal syndrome and corticobasal degeneration: Implications for further study. Journal of Molecular Neuroscience, 45, 350. doi:10.1007/S12031-011-9624-1
- Boeve, B. F., Lang, A. E., & Litvan, I. (2003). Corticobasal degeneration and its relationship to progressive supranuclear palsy and frontotemporal dementia. Annals of Neurology, 54(S5), S15–S19. doi:10.1002/ana.10570
- Borroni, B., Gabirotto, V., Agosti, C., Brambati, S. M., Bellelli, G., Gasparotti, R., … Perani, D. (2008). White matter changes in corticobasal degeneration syndrome and correlation with limb apraxia. Archives of Neurology, 65(6), 796–801. doi:10.1001/archneur.65.6.796
- Borroni, B., Turla, M., Bertasi, V., Agosti, C., Gilberti, N., & Padovani, A. (2008). Cognitive and behavioral assessment in the early stages of neurodegenerative extrapyramidal syndromes. Archives of Gerontology and Geriatrics, 47, 53–61. doi:10.1016/j.archger.2007.07.005
- Buckner, R. L., Snyder, A. Z., Shannon, B. J., LaRossa, G., Sachs, R., Fotenos, A. F., & Mintun, M. A. (2005). Molecular, structural, and functional characterization of alzheimer’s disease: Evidence for a relationship between default activity, amyloid, and memory. Journal of Neuroscience, 25(34), 7709–7717. doi:10.1523/JNEUROSCI.2177-05.2005
- Burrell, J. R., Hodges, J. R., & Rowe, J. B. (2014). Cognition in corticobasal syndrome and progressive supranuclear palsy. A review. Movement Disorders, 29(5), 684–693. doi:10.1002/mds.25872
- Burrell, J. R., Hornberger, M., Villemagne, V. L., Rowe, C. C., & Hodges, J. R. (2013). Clinical profile of PiB-positive corticobasal syndrome. PLoS ONE, 8(4), e61025. doi:10.1371/journal.pone.0061025
- Buxbaum, L. J., Kyle, K., Grossman, M., & Coslett, B. (2007). Left inferior parietal representations for skilled hand-object interactions: Evidence from stroke and corticobasal degeneration. Cortex, 43, 411–423. doi:10.1016/S0010-9452(08)70466-0
- Capasso, R., & Miceli, G. (2001). Esame neuropsicologico per l’Afasia: ENPA (Vol. 4). Springer Science & Business Media.
- Catani, M. (2005). The rises and falls of disconnection syndromes. Brain, 128(10), 2224–2239. doi:10.1093/brain/awh622
- Catani, M., Jones, D. K., & Ffytche, D. H. (2005). Perisylvian language networks of the human brain. Annals of Neurology, 57(1), 8–16. doi:10.1002/ana.20319
- Ceravolo, R., Rossi, C., Cilia, R., Tognoni, G., Antonini, A., Volterrani, D., & Bonuccelli, U. (2013). Evidence of delayed nigrostriatal dysfunction in corticobasal syndrome: A SPECT follow-up study. Parkinsonism & Related Disorders, 19(5), 557–559. doi:10.1016/j.parkreldis.2013.01.013
- Chainay, H., & Humphreys, G. W. (2003). Ideomotor and ideational apraxia in corticobasal degeneration: A case study. Neurocase, 9(2), 177–186. doi:10.1076/neur.9.2.177.15073
- Cilia, R., Rossi, C., Frosini, D., Volterrani, D., Siri, C., Pagni, C., … Ceravolo, R. (2011). Dopamine transporter SPECT imaging in corticobasal syndrome. PLoS ONE, 6(5), e18301. doi:10.1371/journal.pone.0018301
- Clark, D. L., Boutros, N. N., & Mendez, M. F. (2005). The brain and behavior: An introduction to behavioral neuroanatomy. New York, NY: Cambridge University Press.10.1017/CBO9780511543661
- Conson, M., Salzano, S., Manzo, V., Grossi, D., & Trojano, L. (2009). Closing-in without severe drawing disorders: The “fatal” consequences of pathological attraction. Cortex, 45(3), 285–292. doi:10.1016/j.cortex.2007.11.013
- Corbetta, M., & Shulman, G. (2002). Control of goal-directed and stimulus-driven attention in the brain. Nature Reviews Neuroscience, 3, 201–215. doi:10.1038/nrn755
- Crutch, S. J., Schott, J. M., Rabinovici, G. D., Murray, M., Snowden, J. S., van der Flier, W. M., … Boeve, B. F. (2017). Consensus classification of posterior cortical atrophy. Alzheimer’s & Dementia. doi:10.1016/j.jalz.2017.01.014
- Della Sala, S., MacPherson, S. E., Phillips, L. H., Sacco, L., & Spinnler, H. (2003). How many camels are there in Italy? Cognitive estimates standardised on the Italian population. Neurological Sciences, 24(1), 10–15. doi:10.1007/s100720300015
- De Luca, M., Beckmann, C. F., De Stefano, N., Matthews, P. M., & Smith, S. M. (2006). fMRI resting state networks define distinct modes of long-distance interactions in the human brain. NeuroImage, 29(4), 1359–1367. doi:10.1016/j.neuroimage.2005.08.035
- De Renzi, E. (1985). Methods of limb apraxia examination and their bearing on the interpretation of the disorder. In E. A. Roy (Ed.), Neuropsychological studies of apraxia and related disorders (pp. 45–64). Amsterdam: The Netherlands.
- De Renzi, E., Faglioni, P., Scarpa, M., & Crisi, G. (1986). Limb apraxia in patients with damage confined to the left basal ganglia and thalamus. Journal of Neurology, Neurosurgery and Psychiatry, 49, 1030–1038. doi:10.1136/jnnp.49.9.1030
- De Renzi, E., Motti, F., & Nichelli, P. (1980). Imitating gestures. A quantitative approach to ideomotor apraxia. Archives of Neurology, 37, 6–10. doi:10.1001/archneur.1980.00500500036003
- De Renzi, E., Pieczuro, A., & Vignolo, L. A. (1966). Oral apraxia and aphasia. Cortex, 2, 50–73. doi:10.1016/S0010-9452(66)80028-X
- De Renzi, E., Pieczuro, A., & Vignolo, L. A. (1968). Ideational apraxia: A quantitative study. Neuropsychologia, 6(1), 41–52. doi:10.1016/0028-3932(68)90037-7
- Delrieu, J., Payoux, P., Toulza, O., Esquerre, J.-P., Vellas, B., & Voisin, T. (2010). Sensory alien hand syndrome in corticobasal degeneration: A cerebral blood flow study. Movement Disorders, 25, 1288–1291. doi:10.1002/mds.23064
- Denny-Brown, D. (1958). The nature of apraxia. The Journal of Nervous and Mental Disease, 126(1), 9–32. Retrieved from http://journals.lww.com/jonmd/Citation/1958/01000/the_Nature_of_Apraxia.3.aspx10.1097/00005053-195801000-00003
- Di Stefano, F., Kas, A., Habert, M. O., Decazes, P., Lamari, F., Lista, S., … Teichmann, M. (2016). The phenotypical core of Alzheimer’s disease-related and nonrelated variants of the corticobasal syndrome: A systematic clinical, neuropsychological, imaging, and biomarker study. Alzheimer’s & Dementia, 12(7), 786–795. doi:10.1016/j.jalz.2016.02.005
- Dopper, E. G., Seelaar, H., Chiu, W. Z., de Koning, I., van Minkelen, R., Baker, M. C., & Van Swieten, J. C. (2011). Symmetrical corticobasal syndrome caused by a novel c.314dup progranulin mutation. Journal of Molecular Neuroscience, 45(3), 354. doi:10.1007/s12031-011-9626-z
- Ellis, A. W., Flude, B. M., & Young, A. W. (1987). “Neglect dyslexia” and the early visual processing of letters in words and nonwords. Cognitive Neuropsychology, 4(4), 439–464. doi:10.1080/02643298708252047
- FitzGerald, D. B., Drago, V., Jeong, Y., Chang, Y.-L., White, K. D., & Heilman, K. M. (2007). Asymmetrical alien hands in corticobasal degeneration. Movement Disorders, 22(4), 581–584. doi:10.1002/mds.21337
- Folstein, M. F., Folstein, S. E., & McHugh, P. R. (1975). “Mini-mental state”: A practical method for grading the cognitive state of patients for the clinician. Journal of Psychiatric Research, 12(3), 189–198. doi:10.1016/0022-3956(75)90026-6
- Frattali, C. M., Grafman, J., Patronas, N., Makhlouf, F., & Litvan, I. (2000). Language disturbances in corticobasal degeneration. Neurology, 54(4), 990–992. doi:10.1212/WNL.54.4.990
- Gauthier, L., Dehaut, F., & Joanette, Y. (1989). The Bells Test: A quantitative and qualitative test for visual neglect. International Journal of Clinical Neuropsychology, 11(2), 49–54. Retrieved from http://psycnet.apa.org/psycinfo/1989-31545-001
- Giorelli, M., Losignore, N. A., Bagnoli, J., Difazio, P., & Zimatore, G. B. (2014). The progression of posterior cortical atrophy to corticobasal syndrome: Lumping or splitting neurodegenerative diseases? Tremor and Other Hyperkinetic Movements. doi:10.7916/D81G0JCQ
- Goldenberg, G. (2003). Neuropsychological assessment and treatment of disorders of voluntary movements. In P. Halligan, U. Kischka, & J. C. Marshall (Eds.), Handbook of clinical neuropsychology (pp. 340–352). New York: Oxford University Press.
- Goldenberg, G. (2009). Apraxia and the parietal lobes. Neuropsychologia, 47, 1449–1459. doi:10.1016/j.neuropsychologia.2008.07.014
- Goldenberg, G., & Spatt, J. (2009). The neural basis of tool use. Brain, 132, 1645–1655. doi:10.1093/brain/awp080
- Gonzalez Rothi, L. J., Ochipa, C., & Heilman, K. M. (1991). A cognitive neuropsychological model of limb praxis. Cognitive Neuropsychology, 8(6), 443–458. doi:10.1080/02643299108253382
- Graham, N. L., Bak, T. H., & Hodges, J. R. (2003). Corticobasal degeneration as a cognitive disorder. Movement Disorders, 18(11), 1224–1232. doi:10.1002/mds.10536
- Graham, N. L., Zeman, A., Young, A. W., Patterson, K., & Hodges, J. R. (1999). Dyspraxia in a patient with corticobasal degeneration: The role of visual and tactile inputs to action. Journal of Neurology, Neurosurgery and Psychiatry, 67, 334–344. doi:10.1136/jnnp.67.3.334
- Grijalvo-Perez, A. M., & Litvan, I. (2014). Corticobasal degeneration. Seminars in Neurology, 34, 160–173. doi:10.1055/s-0034-1381734
- Guo, J. L., & Lee, V. M. (2014). Cell-to-cell transmission of pathogenic proteins in neurodegenerative diseases. Nature Medicine, 20(2), 130–138. doi:10.1038/nm.3457
- Hammesfahr, S., Antke, C., Mamlins, E., Beu, M., Wojtecki, L., Ferrea, S., … Südmeye, M. (2016). FP-CIT- and IBZM-SPECT in corticobasal syndrome: Results from a clinical follow-up study. Neurodegenerative Diseases, 16(5–6), 342–347. doi:10.1159/000443667
- Hassan, A., Whitwell, J. L., Boeve, B. F., Jack, C. R., Parisi, J. E., Dickson, D. W., & Josephs, K. A. (2010). Symmetric corticobasal degeneration (S-CBD). Parkinsonism & Related Disorders, 16(3), 208–214. doi:10.1016/j.parkreldis.2009.11.013
- Homma, T., Takubo, H., Takahashi, K., Matsubara, S., Takahashi, M., Funata, N., … Uchihara, T. (2014). Lateralized cortical involvement and contralateral parkinsonism without basal ganglia involvement in two autopsy cases of corticobasal syndrome-Alzheimer’s disease. Journal of Alzheimer’s Disease, 40(1), 51–55. doi:10.3233/JAD-131676
- Huang, N., Hornberger, M., Hodges, J. R., & Burrell, J. R. (2014). Measuring disease progression in corticobasal syndrome. Journal of Neurology, 261(8), 1598–1605. doi:10.1007/s00415-014-7389-5
- Hu, W. T., Rippon, G. W., Boeve, B. F., Knopman, D. S., Petersen, R. C., Parisi, J. E., & Josephs, K. A. (2009). Alzheimer’s disease and corticobasal degeneration presenting as corticobasal syndrome. Movement Disorders, 24(9), 1375–1379. doi:10.1002/mds.22574
- Iaccarino, L., Chieffi, S., & Iavarone, A. (2014). Utilization behavior: What is known and what has to be known? Behavioural Neurology, 2014, 1599, Article ID 297128. doi:10.1155/2014/29712
- Jacobs, D. H., Adair, J. C., Macauley, B., Gold, M., Gonzalez Rothi, L. J., & Heilman, K. M. (1999). Apraxia in corticobasal degeneration. Brain and Cognition, 40, 336–354. doi:10.1006/brcg.1999.1085
- Josephs, K. A., Whitwell, J. L., Boeve, B. F., Knopman, D. S., Petersen, R. C., Hu, W. T., … Jack, C. R. (2010). Anatomical differences between CBS-corticobasal degeneration and CBS-Alzheimer’s disease. Movement Disorders, 25(9), 1246–1252. doi:10.1002/mds.23062
- Katz, S., Downs, T. D., Cash, H. R., & Grotz, R. C. (1970). Progress in development of the index of ADL. The Gerontologist, 10(1 Part 1), 20–30. doi:10.1093/geront/10.1_Part_1.20
- Kimura, N., Kumamoto, T., Hanaoka, T., Hazama, Y., Nakamura, K., & Arakawa, R. (2008). Corticobasal de generation presentino with progressive conduction aphasia. Journal of the Neurological Sciences, 269, 163–168. doi:10.1016/j.jns.2007.12.017
- Kloesel, B., Czarnecki, K., Muir, J. J., & Scott Keller, A. (2010). Sequellae of a left-sided parietal stroke: posterior alien hand syndrome. Neurocase, 16(6), 488–493. doi:10.1080/13554794.2010.497154
- Laiacona, M., Barbarotto, R., Trivelli, C., & Capitani, E. (1993). Dissociazioni semantiche intercategoriali: Descrizione di una batteria standardizzata e dati normativi. Archivio di Psicologia, Neurologia e Psichiatria, 54, 209–248. Retrieved from http://hdl.handle.net/2434/194743
- Lang, A. E., Riley, D. E., & Bergeron, C. (1994). Cortico-basal ganglionic degeneration. In D. B. Calne (Ed.), Neurodegenerative diseases. (pp. 877–894). Philadelphia, PA: WB Saunders.
- Lavados, M., Carrasco, X., Pena, M., Zaidel, E., Zaidel, D., & Aboitiz, F. (2002). A new sign of callosal disconnection syndrome: Agonistic dyspraxia. A case study. Neurocase, 8(6), 480–483. doi:10.1076/neur.8.5.480.16178
- Lawton, M. P., & Brody, E. M. (1969). Assessment of older people: Self-maintaining and instrumental activities of daily living. The Gerontologist, 9, 179–186.10.1093/geront/9.3_Part_1.179
- Lee, S. E., Rabinovici, G. D., Mayo, M. C., Wilson, S. M., Seeley, W. W., DeArmond, S. J., … Sidhu, M. (2011). Clinicopathological correlations in corticobasal degeneration. Annals of Neurology, 70(2), 327–340. doi:10.1002/ana.22424
- Leiguarda, R. C., Merello, M., Nouzeilles, M. I., Balej, J., Rivero, A., & Nogués, M. (2003). Limb-kinetic apraxia in corticobasal de generation: clinical and kinematic features. Movement Disorders, 18(1), 49–59. doi:10.1002/mds.10303
- Lheman, S., & Schnider, A. (2008). Memory disorders. In S. F. Cappa (Ed.), Cognitive neurology: A clinical textbook (pp. 119–141). USA: Oxford University Press.10.1093/acprof:oso/9780198569275.001.0001
- Lhermitte, F. (1983). Utilization behaviour and its relation to lesions of the frontal lobes. Brain, 106, 237–255. doi:10.1093/brain/106.2.237
- Lhermitte, F., Pillon, B., & Serdaru, M. (1986). Human autonomy and the frontal lobes. Part I: Imitation and utilization behavior: A neuropsychological study of 75 patients. Annals of Neurology, 19(4), 326–334. doi:10.1002/ana.410190404
- Luzzatti, C., Willmes, K., & De Bleser, R. (1996). Aachener aphasie test: Versione Italiana. Firenze: Organizzazioni Speciali.
- Mahapatra, R. K., Edwards, M. J., Schott, J. M., & Bhatia, K. P. (2004). Corticobasal degeneration. The Lancet Neurology, 3, 736–743. doi:10.1016/S1474-4422(04)00936-6
- Malloy, P. F., Cohen, R. A., & Jenkins, M. A. (1998). Frontal lobe function and dysfunction. In P. J. Snyder, P. D. Nussbaum, & D. L. Robins (Eds.), Clinical neuropsychology: A pocket handbook for assessment (pp. 573–590). Washington, DC: American Psychological Association.
- Mathew, R., Bak, T. H., & Hodges, J. R. (2011). Diagnostic criteria for corticobasal syndrome: A comparative study. Journal of Neurology, Neurosurgery, and Psychiatry. doi:10.1136/jnnp-2011-300875
- McMillan, C. T., Boyd, C., Gross, R. G., Weinstein, J., Firn, K., Toledo, J. B., … Lee, E. B. (2016). Multimodal imaging evidence of pathology-mediated disease distribution in corticobasal syndrome. Neurology, 87(12), 1227–1234. doi:10.1212/WNL.0000000000003119
- McMonagle, P., Blair, M., & Kertesz, A. (2006). Corticobasal degeneration and progressive aphasia. Neurology, 67, 1444–1451. doi:10.1212/01.wnl.0000240215.43492.01
- Mendez, M. F. (2000). Corticobasal ganglionic degeneration with Balint’s syndrome. The Journal of Neuropsychiatry and Clinical Neurosciences, 12, 273–275. doi:10.1176/jnp.12.2.273
- Merians, A. S., Clark, M., Poizner, H., Jacobs, D. H., Adair, J. C., Macauley, B., … Heilman, K. M. (1999). Apraxia differs in corticobasal degeneration and left-parietal stroke: A case study. Brain and Cognition, 40(2), 314–335. doi:10.1006/brcg.1999.1084
- Murray, R., Neumann, M., Forman, M. S., Farmer, J., Massimo, L., Rice, A., … Gorno-Tempini, M. L. (2007). Cognitive and motor assessment in autopsy-proven corticobasal degeneration. Neurology, 68(16), 1274–1283. doi:10.1212/01.wnl.0000259519.78480.c3
- Norman, D. A. (1988). The psychology of everyday things. New York, NY: Basic books.
- Novelli, G., Papagno, C., Capitani, E., Laiacona, M., Vallar, G., & Cappa, S. F. (1986). Three clinical tests for the assessment of lexical retrieval and production: Norms from 320 normal subjects. Archivio di Psicologia, Neurologia e Psichiatria, 47, 477–506.
- Okuda, B., Tanaka, H., Kawabata, K., Tachibana, H., & Sugita, M. (2001). Truncal and limb apraxia in corticobasal degeneration. Movement Disorders, 16, 760–762. doi:10.1002/mds.1150
- Orsini, A., Grossi, D., Capitani, E., Laiacona, M., Papagno, C., & Vallar, G. (1987). Verbal and spatial immediate memory span: Normative data from 1355 adults and 1112 children. The Italian Journal of Neurological Sciences, 8(6), 537–548. doi:10.1007/BF02333660
- Ouchi, H., Toyoshima, Y., Tada, M., Oyake, M., Aida, I., Tomita, I., … M. L., T. (2014). Pathology and sensitivity of current clinical criteria in corticobasal syndrome. Movement Disorders, 29(2), 238–244. doi:10.1002/mds.25746
- Ozsancak, C., Auzou, P., & Dujardin & Hannequin, D. (2000). Dysarthria and orofacial apraxia in corticobasal degeneration. Movement Disorders, 15(5), 905–910. doi:10.1002/1531-8257(200009)15:5<905::AID-MDS1022>3.0.CO;2-D
- Ozsancak, C., Auzou, P., & Hannequin, D. (2004). Orofacial apraxia in corticobasal de generation, progressive supranuclear palsy, multiple system atrophy and Parkinson’s disease. Journal of Neurology, 251(11), 1317–1323. doi:10.1007/s00415-004-0530-0
- Passov, V., Gavrilova, R. H., Strand, E., Cerhan, J. H., & Josephs, K. A. (2011). Sporadic corticobasal syndrome with progranulin mutation presenting as progressive apraxic agraphia. Archives of Neurology, 68(3), 376–380. doi:10.1001/archneurol.2011.26
- Peigneux, P., Salmon, E., Garraux, G., Laureys, S., Willems, S., Dujardin, K., … Franck, G. (2001). Neural and cognitive bases of upper limb apraxia in corticobasal degeneration. Neurology, 57(7), 1259–1268. doi:10.1212/WNL.57.7.1259
- Pharr, V., Uttl, B., Stark, M., Litvan, I., Fantie, B., & Grafman, J. (2001). Comparison of apraxia in corticobasal degeneration and progressive supranuclear palsy. Neurology, 56, 957–963. doi:10.1212/WNL.56.7.957
- Posner, M. I., & Petersen, S. E. (1990). The attention system of the human brain. Annual Review of Neuroscience, 13(1), 25–42. doi:10.1146/annurev.ne.13.030190.000325
- Possin, K. L. (2010). Visual spatial cognition in neurodegenerative disease. Neurocase, 16(6), 466–487. doi:10.1080/13554791003730600
- Rajagopal, R., Bateman, R., & Van Stavern, G. P. (2011). Visual involvement in corticobasal syndrome. Journal of Neuro-Ophthalmology, 1–3. doi:10.1097/WNO.0b013e3182305162
- Riddoch, M. J., Chechlacz, M., Mevorach, C., Mavritsaki, E., Allen, H., & Humphreys, G. W. (2010). The neural mechanisms of visual selection: The view from neuropsychology. Annals of the New York Academy of Sciences, 1191, 156–181. doi:10.1111/j.1749-6632.2010.05448.x
- Riddoch, M. J., & Humphreys, G. W. (1993). BORB: Birmingham object recognition battery. Hove: Lawrence Erlbaum Associates.
- Rittman, T., Ghosh, B., & Rowe, J. (2013). Exploration of functional brain networks in neurodegenerative disease. The Lancet, 381, S92. doi:10.1016/S0140-6736(13)60532-2
- Roy, E. A. (1996). Hand preference, manual asymmetries, and limb apraxia. In D. Elliot (Ed.), Manual asymmetries in motor control (pp. 215–236). Boca Raton, FL: CRC Press.
- Roy, E. A., Square, P. A., Adams, S., & Friesen, H. (1985). Error/movement notation systems in apraxia. Recherches Semitiques/Semiotic Inquiry, 5, 402.
- Rusconi, E., Pinel, P., Dehaene, S., & Kleinschmidt, A. (2010). The enigma of Gerstmann’s syndrome revisited: A telling tale of the vicissitudes of neuropsychology. Brain, 133, 320–332. doi:10.1093/brain/awp281
- Ryan, N. S., Shakespeare, T. J., Lehmann, M., Keihaninejad, S., Nicholas, J. M., Leung, K. K., … Crutch, S. J. (2014). Motor features in posterior cortical atrophy and their imaging correlates. Neurobiology of Aging, 35(12), 2845–2857. doi:10.1016/j.neurobiolaging.2014.05.028
- Salter, J. E., Roy, E. A., Black, S. E., Joshi, A., & Almeida, Q. (2004). Gestural imitation and limb apraxia in corticobasal degeneration. Brain and Cognition, 55, 400–402. doi:10.1016/j.bandc.2004.02.058
- de Schotten, M. T., Dell’Acqua, F., Forkel, S. J., Simmons, A., Vergani, F., Murphy, D. G., & Catani, M. (2011). A lateralized brain network for visuospatial attention. Nature Neuroscience, 14(10), 1245–1246. doi:10.1038/nn.2905
- Seeley, W. W., Crawford, R. K., Zhou, J., Miller, B. L., & Greicius, M. D. (2009). Neurodegenerative diseases target large-scale human brain networks. Neuron, 62, 42–52. doi:10.1016/j.neuron.2009.03.024
- Seeley, W. W., Menon, V., Schatzberg, A. F., Keller, J., Glover, G. H., Kenna, H., … Greicius, M. D. (2007). Dissociable intrinsic connectivity networks for salience processing and executive control. Journal of Neuroscience, 27(9), 2349–2356. doi:10.1523/JNEUROSCI.5587-06.2007
- Semenza, C. (2003). Assessing disorders of awareness and representation of body parts. In P. W. Halligan, U. Kischka, & J. Marshall (Eds.), Handbook of clinical neuropsychology (Chapter 12, pp. 195–213). New York, NY: Oxford University Press.
- Semenza, C., & Goodglass, H. (1985). Localization of body parts in brain injured subjects. Neuropsychologia, 23(2), 161–175. doi:10.1016/0028-3932(85)90101-0
- Shakespeare, T. J., Yong, K. X., Foxe, D., Hodges, J., & Crutch, S. J. (2015). Pronounced impairment of everyday skills and self-care in posterior cortical atrophy. Journal of Alzheimer, 43(2), 381–384. doi:10.3233/JAD-141071
- Soliveri, P., Monza, D., Paridi, D., Radice, D., Grisoli, M., Testa, D., … Girotti, F. (1999). Cognitive and magnetic resonance imaging aspects of corticobasal degeneration and progressive supranuclear palsy. Neurology, 53(3), 502–502. doi:10.1212/WNL.53.3.502
- Soliveri, P., Piacentini, S., & Girotti, F. (2005). Limb apraxia in corticobasal degeneration and progressive supranuclear palsy. Neurology, 64, 448–453. doi:10.1212/01.WNL.0000150732.92567.BA
- Soliveri, P., Piacentini, S., Paridi, D., Testa, D., Carella, F., & Girotti, F. (2003). Distal-proximal differences in limb apraxia in corticobasal degeneration but not progressive supranuclear palsy. Neurological Sciences, 24, 213–214. doi:10.1007/s10072-003-0136-9
- Spatt, J., Bak, T., Bozeat, S., Patterson, K., & Hodges, J. R. (2002). Apraxia, mechanical problem solving and semantic knowledge. Journal of Neurology, 249, 601–608. doi:10.1007/s004150200070
- Spinnler, H., & Tognoni, G. (1987). Standardizzazione e taratura italiana di test neuropsicologici. Italian Journal of Neurological Sciences, 8(Supplement 8), 1–120.
- Stamenova, V., Roy, E. A., & Black, S. (2009). A model-based approach to understanding apraxia in corticobasal syndrome. Neuropsychology Review, 19, 47–63. doi:10.1007/s11065-008-9079-5
- Stamenova, V., Roy, E. A., & Black, S. E. (2011). Limb apraxia in corticobasal syndrome. Cortex, 47, 460–472. doi:10.1016/j.cortex.2010.04.010
- Südmeyer, M., Pieperhoff, P., Ferrea, S., Krause, H., Groiss, S. J., Elben, S., … Schnitzler, A. (2012). Longitudinal deformation-based morphometry reveals spatio-temporal dynamics of brain volume changes in patients with corticobasal syndrome. PLoS ONE, 7(7), e41873. doi:10.1371/journal.pone.0041873
- Sunderland, A., & Shinner, C. (2007). Ideomotor apraxia and functional ability. Cortex, 43, 359–367. doi:10.1016/S0010-9452(08)70461-1
- Tang-Wai, D. F., Josephs, K. A., Boeve, B. F., Dickson, D. W., Parisi, J. E., & Petersen, R. C. (2003). Pathologically confirmed corticobasal degeneration presenting with visuospatial dysfunction. Neurology, 61, 1134–1135. doi:10.1212/01.WNL.0000086814.35352.B3
- Tree, J., & Kay, J. (2008). Longitudinal assessment of language and memory impairments in pathologically confirmed cortico-basal ganglionic degeneration. Cortex, 44, 1234–1247. doi:10.1016/j.cortex.2007.08.017
- Vallar, G. (2007). Spatial neglect, Balint-Holmes’ and Gerstmann’s syndromes, and other spatial disorders. CNS Spectrums, 12(7), 527–536. doi:10.1017/S1092852900021271
- Warrington, E. K., & James, M. (1991). The VISUAL object and space perception battery. Bury St. Edmunds: Thames Valley Test Company.
- Weintraub, S., & Mesulam, M. (2009). Scientific commentary. With or without FUS, it is the anatomy that dictates the dementia phenotype. Brain, 132, 2906–2908. doi:10.1093/brain/awp286
- Whitwell, J. L., Jack, C. R., Boeve, B. F., Parisi, J. E., Ahlskog, J. E., Drubach, D. A., … Josephs, K. A. (2010). Imaging correlates of pathology in corticobasal syndrome. Neurology, 75(21), 1879–1887. doi:10.1212/WNL.0b013e3181feb2e8
- Wolpe, N., Moore, J. W., Rae, C. L., Rittman, T., Altena, E., Haggard, P., & Rowe, J. B. (2014). The medial frontal-prefrontal network for altered awareness and control of action in corticobasal syndrome. Brain, 137, 208–220. doi:10.1093/brain/awt302