
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Molecular parameters such as equilibrium structure, dipole moment, rotational constant, harmonic frequency, IR intensity, adiabatic electron affinity, atomisation energy and ionisation potential of some P-bearing molecules PS, PO and HC3P in their neutral, cationic and anionic forms were investigated using the popular B3LYP hybrid density functional with four basis sets 6-311++G(2df,2pd), 6-311++G(3df,3pd), cc-pVTZ and aug-cc-pVTZ. The computed data conform well to those existing in the literature. Therefore, the predicted data for those molecules or ions which are not available in the literature should be reliable.
Public Interest Statement
Structural and spectroscopic parameters of molecules are usually obtained through experimental methods. However, it is possible to use highly accurate computational methods for predicting these parameters. We have shown that there is a good comparison between the experimental parameters and those predicted by computational methods for some astrophysical phosphorus containing species. Our work should therefore be useful for those phosphorus species where structural and spectroscopic parameters are not reported.
1. Introduction
Agúndez, Cernicharo, and Guélin (Citation2007) detected phosphaethyne (HCP) in the AGB star envelope IRC +10216. According to chemical models performed by Agúndez et al. (Citation2007), other P-bearing molecules that are likely to be detected are phosphorus monosulphide (PS) and phosphorus monoxide (PO) in O-rich circumstellar envelopes and phosphabutadiyne (HC3P) in C-rich circumstellar envelopes. The identification of HC3P is highly probable due to the reaction between carbon monophosphide (CP) and acetylene (C2H2), , studied theoretically by Yu, Zhao, Kan, and Fu (Citation2006) using B3LYP and QCSID(T) methods.
Interstellar grain mass is mostly formed by accretion-a process directly related to the depletion of elements. Phosphorus is generally believed to be highly depleted in dense molecular clouds. As pointed out by Agúndez et al. (Citation2007), phosphorus should be present in the hot stellar atmospheres of late-stage stars and thermochemical equilibrium calculations suggest that it is locked into PS and PO (O-rich envelopes) or HCP (C-rich envelopes). When a cold envelope is formed through the expansion of the atmospheric gas, most of the heavy elements condense on dust grains. The observation and study of the P-bearing molecules are thereby important for understanding the chemistry of grains in interstellar/circumstellar medium.
Experimental work on the equilibrium structures of PO and PS molecules involves spectroscopic investigation by Huber and Herzberg (Citation1979). The electron affinity (EA) of PO was studied by Brinkmann, Tschumper, and Schaefer (Citation1999) where they employed six functionals. They also rendered values for the optimised structures and harmonic frequencies of PO and its anion PO−. Zittel and Lineberger (Citation1976) utilised a fixed-frequency laser photoelectron spectroscopy to study beams of PO− produced from phosphine burned with N2O in a low-pressure discharge source. They obtained an EA of 1.092 ± 0.010 eV for PO molecule. Dyke, Morris, and Ridha (Citation1982) studied the ground state of PO+ by means of a vacuum ultraviolet photoelectron spectroscopy and the adiabatic ionisation potential (IP) of PO was measured as 8.39 ± 0.01 eV. The G2 value determined by Curtiss, Raghavachari, Trucks, and Pople (Citation1991) for the atomisation energy (AE) of PO is 98.9 kcal/mol. Metropoulos, Papakondylis, and Mavridis (Citation2003) computed the accurate potential energy curves of the ground states of the PO, PO+ and PO− species using multireference method. Müller and Woon (Citation2013) calculated the dipole moments for silicon and phosphorus compounds of astrophysical interest.
Botschwina, Oswald, Linnartz, and Verdes (Citation2000) reported the equilibrium geometry, harmonic vibrational wavenumbers, vibration–rotation coupling constant and l-type doubling constants of HC3P molecule using CCSD(T) method. The only experimental investigation of HC3P is the study of the microwave spectrum by Kroto, Nixon, and Ohno (Citation1981), from which rotational, quartic centrifugal distortions and l-type doubling constants were determined. No experimental and theoretical findings are available for the anion and cation of HC3P molecule.
This theoretical research targets some identified and potential (P-bearing) interstellar molecules using the B3LYP functional and four basis sets. The objectives of this work were: (1) to assess the performance of the functional and the basis sets to calculate structural, spectroscopic and energetic parameters and (2) to ascertain the computed parameters for those not yet available in the literature.
2. Methodology
The hybrid density functional B3LYP was employed to provide insight into the molecular properties of the potential P-bearing interstellar molecules. Computations were performed using four different basis sets 6-311++G(2df,2pd), 6-311++G(3df,3pd), cc-pVTZ and aug-cc-pVTZ (Clark, Chandrasekhar, Spitznagel, & Schleyer, Citation1983; Dunning, Citation1989; Frisch, Pople, & Binkley, Citation1984; Gill, Johnson, Pople, & Frisch, Citation1992; Kendall, Dunning, & Harrison, Citation1992; Woon & Dunning, Citation1993). For each basis set, the molecular structures were optimised under tight convergence criteria and frequency computations were carried out to verify the nature of the stationary points. Similar procedures were applied to the anion and cation of each species. All computations were performed using a suite of Gaussian 03W programs (Frisch et al., Citation2003).
The adiabatic EA, EA = E(optimised neutral) − E(optimised anion), of each molecule was computed as the difference between the total energy of the optimised neutral molecule and the total energy of the corresponding optimised anion.
The AE was calculated as the energy difference between the optimised neutral molecule and its corresponding constituent atoms. Adiabatic IP is the energy necessary to remove an electron from the outermost filled molecular orbital of the ground state.
The adiabatic IP, IP = E(optimised neutral) − E(optimised cation), was computed as the difference between the total energy of the optimised neutral molecule and the total energy of the corresponding optimised cation.
3. Results and discussion
3.1. Equilibrium geometries of potential P-bearing astromolecules
The computed equilibrium bond lengths of the two P-bearing diatomic molecules with their anions and cations are shown in Table along with empirical and earlier theoretical data. The 6-311++G(3df,3pd) basis set produces lower values compared to the other three basis sets. The cc-pVTZ and aug-cc-pVTZ basis sets rendered similar values for the studied phosphorus-bearing species except for PS− and PO−. For PS molecule, the predicted 6-311++G(3df,3pd) value is quite close to the experimental value of Huber and Herzberg (Citation1979) while the 6-311++G(2df,2pd) value of PS deviates by about 0.006 Å. The deviation of the remaining computed values using the other three basis sets is 0.011 Å. Good agreement is observed between the computed 6-311++G(3df,3pd) value of PO and the experimental value (Huber & Herzberg, Citation1979) with deviation of 0.002 Å. The cc-pVTZ and aug-cc-pVTZ values of PO are nevertheless in better agreement with the theoretical result of Brinkmann et al. (Citation1999). For PO−, the aug-cc-pVTZ basis set produces a value that remarkably concurs with the experimental result, and hence predicts a much better value than that computed by Brinkmann et al. (Citation1999). Deviation is comparatively significant (about 0.02 Å) for PO− using the cc-pVTZ basis set with reference to the experimental value. It is noted that cc-pVTZ value of PO− differs quite considerably to the values obtained from the other three basis sets.
Table 1. Bond lengths (Å) of PS, PO, and their anions and cations
Table illustrates the optimised structural parameters of HC3P, HC3P− and HC3P+. Contrary to HC3P and HC3P+, a non-linear minima was located for HC3P− (Figure ) using the four basis sets.
Table 2. Equilibrium geometries of HC3P, HC3P− and HC3P+
Figure 1. Equilibrium geometry of HC3P−.
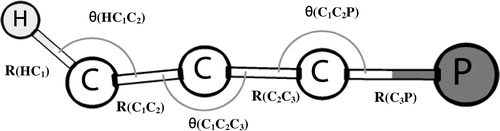
The computed structural parameters of HC3P agree quite well with the CCSD(T)/cc-pVTZ result of Botschwina et al. (Citation2000). The 6-311++G(2df,2pd) and aug-cc-pVTZ basis sets produce values of R(HC1) that are identical to the corresponding estimated value of Botschwina et al. (Citation2000) while the other two basis sets underestimate by only 0.001 Å. For the four basis sets, the calculated values of R(C1C2) are similar and deviate from the corresponding CCSD(T) result by 0.003 Å. A similar situation is observed for the predicted values of R(C2C3) but with a deviation of 0.012 Å with its corresponding CCSD(T) result. For the structural parameter R(C3P), the 6-311++G(2df,2pd) value is in better agreement with the CCSD(T) result than the computed values at the other three basis sets. No experimental and earlier theoretical findings are available for HC3P− and HC3P+ ions. The optimised geometries of the anionic and cationic species are hence reported for the first time. The computed structural parameters of HC3P− do not differ significantly from each other using the four basis sets. For the bond angles θ(HC1C2) and θ(C1C2P), the predicted cc-pVTZ values are however, found to be lower than the values using the other three basis sets.
3.2. Dipole moments of potential P-bearing astromolecules
Table displays the results of the computed dipole moments of the P-bearing species. Experimental and earlier estimated values are only available for HC3P molecule. The aug-cc-pVTZ value is closer with the CCSD(T)/cc-pVTZ value of Botschwina et al. (Citation2000). Nevertheless, the aug-cc-pVTZ value differs by 0.125 D from the dipole moment obtained by Kroto et al. (Citation1981). The 6-311++G(3df,3pd) produces a value that deviates less from the experimental dipole moment of HC3P compared to the predictions of the other three basis sets. For PS− molecule, the cc-pVTZ value is particularly lower than the other computed dipole moments. Also, it is noted that the calculated dipole moments of PO− molecule increase quite significantly with the enhancement of the basis set from 6-311++G(2df,2pd) to aug-cc-pVTZ.
Table 3. Dipole moments (Debye) of the P-bearing molecules
3.3. Rotational constants of potential P-bearing astromolecules
The computed rotational constants of the P-bearing species are compiled in Table . For PS, the 6-311++G(3df,3pd) basis set renders values that conform very well with the observed value (Huber & Herzberg, Citation1979) with a deviation of only 0.002 GHz. For PO molecule, the 6-311++G(3df,3pd) value deviates from the corresponding empirical value (Huber & Herzberg, Citation1979) by 0.047 GHz while the deviations for the other calculated values are more significant. The computed values of HC3P deviate with less than 0.03 GHz from the experimental value (Kroto et al., Citation1981) as well as the estimated value of Botschwina et al. (Citation2000). The cc-pVTZ value of HC3P− is quite low compared to the predicted values using the other three basis sets. For the remaining molecules, no experimental and earlier theoretical data are available for comparison.
Table 4. Rotational constants (GHz) of the P-bearing molecules
3.4. Harmonic frequencies and IR intensities of potential P-bearing astromolecules
Table shows the calculated harmonic frequencies and IR intensities (in parenthesis) of PS, PO, and their respective anions and cations. The cc-pVTZ and aug-cc-pVTZ values of PS concord very well with the experimental value (Huber & Herzberg, Citation1979) compared the other computed values using the other two basis sets. The calculated harmonic frequencies of PO deviate from the experimental value (Huber & Herzberg, Citation1979) by less than 26 cm−1. The aug-cc-pVTZ value of PO is nevertheless closer to the theoretical value of Brinkmann et al. (Citation1999) than the experimental result. For PO−, all the computed harmonic frequencies fall within the error bar of the experimental value. The trend in the computed values of all molecules is their decrease from 6-311++G(3df,3pd) to aug-cc-pVTZ.
Table 5. Harmonic frequencies (cm−1) and IR intensities (km/mol) of PS, PO, and their anions and cations
The calculated harmonic frequencies of HC3P are given in Table with the IR intensities in parenthesis. The computations of the harmonic frequencies of HC3P agree very well with the coupled cluster calculation of Botschwina et al. (Citation2000). Good agreement is observed for the calculated bending frequencies ν5 with the experimental value, nearly all the estimated values fall within the experimental error bar. It is to be noted that the computed bending frequencies ν7 concur well with the experimental result.
Table 6. Harmonic frequencies (cm−1) and IR intensities (km/mol) of HC3P
For HC3P− and HC3P+, the computed harmonic frequencies together with the IR intensities (in parenthesis) are presented in Tables and . There are no experimental and earlier theoretical data available for comparison. The B3LYP approach using each of the four basis sets, all fail to produce degenerate frequencies for the bending modes for HC3P+.
Table 7. Harmonic frequencies (cm−1) and IR intensities (km/mol) of HC3P−
Table 8. Harmonic frequencies (cm−1) and IR intensities (km/mol) of HC3P+
3.5. Molecular energetics of potential P-bearing astromolecules
The electron affinities, atomisation energies and IPs of the P-bearing molecules are shown in Table . For PS molecule, the calculated atomisation energies are quite in good agreement with theoretical value (Curtiss et al., Citation1991) while the computed IPs are found to be smaller than the corresponding experimental value (Drowart, Myers, Szwarc, Vander Auwera-Mahieu, & Uy, Citation1973) by about 10%. For PO molecule, the aug-cc-pVTZ value of the EA is quite comparable to the estimated value of Brinkmann et al. (Citation1999) but large deviations are however noticed for all its computed electron affinities from the experimental result (Zittel & Lineberger, Citation1976). Good agreements are also observed for the predicted cc-pVTZ AE of PO with the G2 result of Curtiss et al. (Citation1991) and for the aug-cc-pVTZ AE of PO with the corresponding observed value. In general, all the computed cc-pVTZ electron affinities are comparatively smaller than the electron affinities predicted at the other three basis sets. No experimental and earlier theoretical data are available for comparison with the computed EA, AE and IP of HC3P.
Table 9. Electron affinities (EA), atomisation energies (AE) and ionisation potentials (IP) in kcal/mol of the P-bearing molecules
4. Conclusions
The computed parameters agree quite well with structural information from experimental and earlier theoretical data wherever available. For molecules or ions not previously studied, the computed data should be reliable. All the four basis sets predict a non-linear minimum configuration for HC3P−. For P-bearing molecules with exception to PS−, PO−, HC3P and HC3P+, the computed values decreases in general from 6-311++G(2df,2pd) to the aug-cc-pVTZ. The estimated rotational constants for P-bearing molecules decrease from 6-311++G(3df,3pd) to aug-cc-pVTZ. The computed electron affinities as well as the computed atomisation energies are reported for the first time for P-bearing compounds PS, HC3P. The trend in most computed values for P-bearing species is their decrease from 6-311++G(3df,3pd) to aug-cc-pVTZ. In general, the computed IPs decrease slightly from 6-311++G(2df,2pd) to aug-cc-pVTZ for the P-bearing species.
Corrigendum
This article was originally published with errors. This version has been corrected. Please see Corrigendum (http://dx.doi.org/10.1080/23311940.2015.1090261).
Cover image
Source: Authors.
Acknowledgements
The authors acknowledge the facilities from their respective universities. The authors also acknowledge the comments from the anonymous reviewers to improve the manuscript.
Additional information
Funding
Notes on contributors
Ponnadurai Ramasami
Professor Ponnadurai Ramasami leads the Computational Chemistry Group at the University of Mauritius. The group is focused towards the use of computational methods to solve chemistry and interdisciplinary problems. This astrophysical project is a good example of multidisciplinary collaboration between University of Mauritius, Mauritius and King Saud University, Saudi Arabia.
References
- Agúndez, M., Cernicharo, J., & Guélin, M. (2007). Discovery of phosphaethyne(HCP) in space: Phosphorus chemistry in circumstellar envelopes. The Astrophysical Journal, 662, L91–L94. Retrieved from http://iopscience.iop.org/1538-4357/662/2/L9110.1086/519561
- Botschwina, P., Oswald, R., Linnartz, H., & Verdes, D. (2000). The ν1 and ν2 bands of Ar HN2+: A joint theoretical/experimental study. The Journal of Chemical Physics, 113, 2736–2740. Retrieved from http://scitation.aip.org/content/aip/journal/jcp/113/7/10.1063/1.130526310.1063/1.1305263
- Brinkmann, N. R., Tschumper, G. S., & Schaefer, H. F. (1999). Electron affinities of the oxides of aluminum, silicon, phosphorus, sulfur, and chlorine. The Journal of Chemical Physics, 110, 6240–6245. Retrieved from http://www.researchgate.net/publication/6500864_Kinetics_of_sulfur_oxide_sulfur_fluoride_and_sulfur_oxyfluoride_anions_with_atomic_species_at_298_and_500_K10.1063/1.478528
- Clark, T., Chandrasekhar, J., Spitznagel, G. W., & Schleyer, P. V. R. (1983). Efficient diffuse function-augmented basis sets for anion calculations. III. The 3-21+G basis set for first-row elements, Li–F. Journal of Computational Chemistry, 4, 294–301. Retrieved from http://onlinelibrary.wiley.com/doi/10.1002/jcc.540040303/abstract10.1002/(ISSN)1096-987X
- Curtiss, L. A., Raghavachari, K., Trucks, G. W., & Pople, J. A. (1991). Gaussian-2 theory for molecular energies of first- and second-row compounds. The Journal of Chemical Physics, 94, 7221–7230. Retrieved from http://www.researchgate.net/publication/234983938_Gaussian2_theory_using_reduced_MllerPlesset_orders10.1063/1.460205
- Drowart, J., Myers, C. E., Szwarc, R., Vander Auwera-Mahieu, A., & Uy, O. M. (1973). The dissociation energies of the molecules PS, PSe, and PTe. High Temperature Science, 5, 482–488.
- Dunning, T. H. (1989). Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. The Journal of Chemical Physics, 90, 1007–1023. Retrieved from http://scitation.aip.org/content/aip/journal/jcp/90/2/10.1063/1.45615310.1063/1.456153
- Dyke, J. M., Morris, A., & Ridha, A. (1982). Study of the ground state of PO+ using photoelectron spectroscopy. Journal of the Chemical Society, Faraday Transactions 2, 78, 2077–2082. Retrieved from http://pubs.rsc.org/en/Content/ArticleLanding/1982/F2/f29827802077#!divAbstract10.1039/f29827802077
- Frisch, M. J., Pople, J. A., & Binkley, J. S. (1984). Self-consistent molecular orbital methods 25. Supplementary functions for Gaussian basis sets. The Journal of Chemical Physics, 80, 3265–3269. Retrieved from http://scitation.aip.org/content/aip/journal/jcp/80/7/10.1063/1.44707910.1063/1.447079
- Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., … Pople, J. A. (2003). Gaussian 03, revision B.03. Pittsburgh, PA: Gaussian.
- Gill, P. M. W., Johnson, B. G., Pople, J. A., & Frisch, M. (1992). The performance of the Becke–Lee–Yang–Parr (B–LYP) density functional theory with various basis sets. Chemical Physics Letters, 197, 499–505. Retrieved from http://www.sciencedirect.com/science/article/pii/000926149285807M10.1016/0009-2614(92)85807-M
- Huber, K. P., & Herzberg, G. (1979). Molecular spectra and molecular structure. Van Nostrand Reinhold. Retrieved from http://link.springer.com/chapter/10.1007%2F978-1-4757-0961-2_210.1007/978-1-4757-0961-2
- Kendall, R. A., Dunning, T. H., & Harrison, R. J. (1992). Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. The Journal of Chemical Physics, 96, 6796–6806. Retrieved from http://scitation.aip.org/content/aip/journal/jcp/96/9/10.1063/1.46256910.1063/1.462569
- Kroto, H. W., Nixon, J. F., & Ohno, K. (1981). The microwave spectrum of phosphabutadiyne, H–C–C–C–P. Journal of Molecular Spectroscopy, 90, 512–516. Retrieved from http://www.sciencedirect.com/science/article/pii/002228528190134X10.1016/0022-2852(81)90143-0
- Metropoulos, A., Papakondylis, A., & Mavridis, A. (2003). Ab initio investigation of the ground state properties of PO, PO+ and PO−. The Journal of Chemical Physics, 119, 5981–5987. Retrieved from http://scitation.aip.org/content/aip/journal/jcp/119/12/10.1063/1.159934110.1063/1.1599341
- Müller, H. S. P., & Woon, D. E. (2013). Calculated dipole moments for silicon and phosphorus compounds of astrophysical interest. The Journal of Physical Chemistry A, 117, 13868–13877. Retrieved from http://pubs.acs.org/doi/abs/10.1021/jp408380710.1021/jp4083807
- Woon, D. E., & Dunning, T. H. (1993). Gaussian basis sets for use in correlated molecular calculations. III. The atoms aluminum through argon. The Journal of Chemical Physics, 98, 1358–1371. Retrieved from http://scitation.aip.org/content/aip/journal/jcp/98/2/10.1063/1.46430310.1063/1.464303
- Yu, H. T., Zhao, Y. L., Kan, W., & Fu, H. G. (2006). A theoretical study on the radical–neutral reaction mechanism of carbon monophosphide, CP, with acetylene, C2H2. Journal of Molecular Structure: THEOCHEM, 772, 45–50. Retrieved from http://www.sciencedirect.com/science/article/pii/S016612800600373310.1016/j.theochem.2006.06.017
- Zittel, P. F., & Lineberger, W. C. (1976). Laser photoelectron spectrometry of PO−, PH−, and PH−2. The Journal of Chemical Physics, 65, 1236–1243. Retrieved from http://scitation.aip.org/content/aip/journal/jcp/65/4/10.1063/1.43323210.1063/1.433232