
Abstract
Poly(amido amine) (PAMAM) dendrimers have been considered as possible delivery systems for anticancer drugs. One potential advantage of these carriers would be their use in oral formulations, which will require absorption in the intestinal lumen. This may require the opening of tight junctions which may be enabled by reducing the Ca2+ concentration in the intestinal lumen, which has been shown as an absorption mechanism for EDTA (ethylenediaminetetraacetic acid). Using molecular dynamics simulations, we show that the G3.5 PAMAM dendrimers are able to chelate Ca2+ at similar proportions to EDTA, providing support to the hypothesis that oral formulations of PAMAM dendrimers could use this high chelating efficiency as a potential mechanism for permeating the tight junctions of the intestines if other formulation barriers could be overcome.
Public Interest Statement
The work presented here shows how computer simulations can aid in the development of new ways to deliver anticancer drugs. In particular, the results presented here demonstrate that the use of PAMAM nanoparticles is a viable approach to deliver cancer treatment drugs by oral formulations. Such formulations can be much more convenient for patients and may also facilitate formulation of drugs that are not suitable for infusion.
1. Introduction
Poly(amido amine) (PAMAM) dendrimers are complex molecules for which their biochemical activity in vivo is not fully understood. A particular mechanism of interest is the pathway by which orally taken PAMAM dendrimers could reach their target location when used as nanocarriers of anticancer drugs. Specifically, there is interest in understanding how these particles would be able to permeate the tight junctions of the intestinal lumen (Wang et al., Citation2010).
Literature consensus shows that tight junctions are dependent upon extracellular calcium (Ca2+) and magnesium for their integrity and function (Noach, Kurosaki, Blom-Roosemalen, & de Boer, Citation1993). Extracellular Ca2+ is responsible for keeping the tight junctions closed and it is known that lower concentrations of Ca2+ in the intestinal lumen lead to their opening. This has been clearly established using ethylenediaminetetraacetic acid (EDTA, Supplementary material Schema S1), a known Ca2+ chelator, that has been shown to open and transverse the tight junctions (Amsterdam & Jamieson, Citation1974). Several publications have suggested that PAMAM dendrimers also would be able to travel across the intestinal barrier using the same mechanism (Hubbard, Ghandehari, & Brayden, Citation2014; Hubbard et al., Citation2015). Due to their anionic charge the carboxylic acid (COOH) terminated Generation 3.5 (G3.5) PAMAM dendrimers may be capable of chelating Ca2+ in solution with a chelation efficiency similar to EDTA (Noach et al., Citation1993). Each COOH terminated G3.5 PAMAM dendrimer (Supplementary material Schema S2) can theoretically chelate 32 Ca2+ ions, thus significantly reducing the concentration of extracellular Ca2+ and therefore creating an extracellular environment that is prone to opening the tight junctions. This could allow for paracellular transport of G3.5 PAMAM dendrimers via the tight junctions. However, the ability of PAMAM dendrimers to chelate Ca2+ has yet to be confirmed by in vitro or in vivo studies. Simulation studies can be used to better understand this hypothesis and justify more experimental work.
Molecular dynamics (MD) simulations are routinely used to provide understanding and testing of novel hypotheses at the molecular scale for small molecule drug delivery research (Shinoda, DeVane, & Klein, Citation2012; Xiang & Anderson, Citation2006). With recent advances in computational power such simulations can now be used in nanomedicine, for instance, to better understand chemical and biological properties of PAMAM dendrimer nanoparticles. Many studies have focused on the use of MD simulations to gain insight on PAMAM dendrimer/ligand conformations and energies. A few molecular docking studies have been reported using MD simulations to analyze the interactions of siRNA and PAMAM dendrimers (Jensen et al., Citation2010; Karatasos, Posocco, Laurini, & Pricl, Citation2012; Vasumathi & Maiti, Citation2010). Other molecular docking studies have involved PAMAM dendrimers and ligands such as curcumin and porphyrin (Cao, Zhang, Wang, Yang, & Jiang, Citation2013; Castriciano et al., Citation2011). These include work by Avila-Salas et al. (Citation2012) who used MD simulations and QSAR methods for in silico dendrimer–drug affinity studies. Ivanov and Jacobson (Citation2008) used MD simulations to test the theoretical possibility of bivalent binding of a dendrimer, covalently appended with multiple copies of a ligand. Lee, Majoros, Williams, Athey, and Baker (Citation2009) used MD simulations along with chemical analysis to guide the design of a multifunctional PAMAM dendrimer-based nanotherapeutic. Barata et al. (Citation2011) used MD simulations to identify the key structural design principles for bioactive dendrimer molecules that could be synthesized and biologically evaluated. MD studies of PAMAM dendrimers chelation by various ions have been also recently reported (Astachov et al., Citation2016; Garzoni, Cheval, Fahmi, Danani, & Pavan, Citation2012), but to the authors’ knowledge no study has been performed on the chelating efficiency of Ca2+ by PAMAM dendrimers. In this article, we demonstrate how MD simulations can be used to test the chelating efficiency of G3.5 PAMAM dendrimers.
2. Methods
EDTA and G3.5 PAMAM dendrimer structures were manually constructed using MarvinSketch by ChemAxon (Citation2013) and ChemAxon, Berry, and Ruyts (Citation2012). The structures for each molecule were converted to their ionic forms by removing a hydrogen atom from the terminal COOH groups. This resulted in a minus four charge for the EDTA molecule, as there are four terminal COOH groups in it, and a minus 64 charge for the G3.5 PAMAM dendrimer molecule where there are 64 terminal COOH groups.
For all simulations, the ff12sb force fields were used along with the general AMBER force field (GAFF) to represent the EDTA and G3.5 PAMAM dendrimer (Case et al., Citation2014; Wang, Wang, Kollman, & Case, Citation2006). Four different solvent boxes were prepared for four different simulations: EDTA with Ca2+ in water, G3.5 PAMAM dendrimer with Ca2+ in water, EDTA with Ca2+ in a buffer solution, and G3.5 PAMAM with Ca2+ in the same buffer solution. The water model used in all the simulations was the TIP3P (Jorgensen, Chandrasekhar, Madura, Impey, & Klein, Citation1983). For both simulation studies in water, the concentration of Ca2+ was adjusted to approximately 0.115 M, while the simulation studies in the buffer solution were done using concentrations of Ca2+ ions of approximately 0.0575 M with the addition of sodium chloride (NaCl) at a concentration of approximately 0.115 M. The concentrations were calculated with respect to the volume of the simulation box and those used in the buffer simulations are representative of the experimental solutions used to examine the ability of EDTA to chelate Ca2+ and increase epithelial absorption by Vllasaliu et al. (Citation2012) and Tomita, Hayashi, and Awazu (Citation1996). The simulations in the buffer solution have been performed to further test the ability of Ca2+ chelation by EDTA and G3.5 PAMAM dendrimers, when a competing ion (Na+) is present in the solution. For the EDTA simulations the volume of the water box was approximately 75,000 Å3, while for the simulations with G3.5 PAMAM dendrimer the volume of the water box was approximately 460,000 Å3. All the simulations were performed using periodic boundary conditions and a non-bonded cutoff of 9.0 Å in the NPT ensemble. Specifically, the following number of molecules were used in the simulations: the EDTA and Ca2+ in water MD simulation has 1 EDTA molecule, 6 Cl− ions, 5 Ca2+ ions, and 2,141 water molecules; the EDTA and Ca2+ in a buffer MD simulation has 1 EDTA molecule, 6 Na+ ions, 6 Cl− ions, 2 Ca2+ ions, and 2,220 water molecules; the G3.5 PAMAM dendrimer and Ca2+ in water MD simulation has 1 G3.5 PAMAM dendrimer molecule, 32 Ca2+ ions, and 13,297 water molecules; the G3.5 PAMAM dendrimer and Ca2+ in a buffer MD simulation has 1 G3.5 PAMAM dendrimer molecule, 32 Na+ ions, 16 Ca2+ ions, and 13,035 water molecules.
Three independent MD simulations were performed for each system to obtain ensemble averages of multiple MD runs, which are those presented in the paper. The corresponding results for each of the simulations are provided in the supplementary material. Before each simulation all of the counter ions were randomly distributed in the water box at 8.00 Å from the molecule of interest, EDTA or G3.5 PAMAM dendrimer, using cpptraj (Roe & Cheatham, Citation2013).
The initial minimization step consisted of a total of 3,000 minimization iterations, of which 1,000 were done using the steepest descent method and 2,000 using conjugate gradients. Following the minimization, we performed a 40-ps equilibration phase where the temperature was increased from 0.0 to 300.0 K using 20,000 steps with a time step of 2 fs and using Langevin dynamics with a collision frequency of 1.0 ps−1. Following the equilibration, we performed a 30-ns MD simulation consisting of 1.5 × 107 steps with a time step of 2 fs. During the MD simulation the temperature and pressure were held constant at 300.0 K and 1.0 bar, respectively, and Langevin dynamics was used with a collision frequency of 1.0 ps−1. The 30-ns MD simulation time was selected according to literature precedence (Vasumathi & Maiti, Citation2010) and the appropriateness of the selection was verified by the observation of the desired behavior (binding events strongly suggesting chelation) within the time frame, so no further simulation was necessary. Furthermore, comparison of the radial distributions at increasingly longer windows shows no significant differences (Supplementary material Figure S5).
Trajectory analysis of the 30-ns MD simulation was performed using cpptraj (Roe & Cheatham, Citation2013). The water molecules were removed from the trajectories and a custom Python script was used to plot the distance from each counter ion to the van der Waals surface of the molecule of interest (EDTA or G3.5 PAMAM dendrimer) for each time step of the 30-ns MD simulation. This script also calculated the average distance and minimum distance of each counter ion to the surface of the molecule of interest (EDTA or G3.5 PAMAM dendrimer) during the 30-ns MD simulation and the percentage of dwell time, that is the number of steps in which an individual counter ion is within 3.0 Å of the surface of the molecule divided by the total number of steps in the MD simulation.
The radial distribution functions were calculated within cpptraj for each set of counter ions (Ca2+, Na+, and Cl−) within each run of the MD simulations. The results reported here correspond to the average radial distributions over the three simulation runs performed for each system considered here (EDTA with Ca2+ in water, G3.5 PAMAM dendrimer with Ca+2 in water, EDTA with Ca2+ in a buffer solution, and G3.5 PAMAM with Ca2+ in the same buffer solution).
All calculations were performed on the Tangent cluster at the Center for High Performance Computing. Tangent is a part of the Adaptable Profile-Driven Testbed (Apt), an initiative by the Flux Research Group to provide a flexible, on-demand cloud computing environment targeted at researchers and scientists (https://www.flux.utah.edu/project/apt). We used our research project as a demonstration on how to use a computationally intensive application in a cloud computing environment. This study shows that it is possible to perform computationally significant drug delivery research in such an environment, which may provide researchers an appealing and more cost-effective alternative to traditional, dedicated high-performance computing (HPC) environments.
3. Results and discussion
As discussed above four different simulations were carried out: EDTA and Ca2+ in water, G3.5 PAMAM dendrimer and Ca2+ in water, EDTA and Ca2+ in a buffer, and G3.5 PAMAM dendrimer and Ca2+ in the same buffer. The results from each of these simulations as well as the performance of the Apt computational environment are described in the following subsections. In all cases our results are reported as an average of the values obtained in each of the three independent MD simulations performed for each system, to represent ensemble average values.
3.1. MD simulation study of EDTA and Ca2+ in water
Results from the EDTA and Ca2+ in water MD simulations are presented in Figure and Table . Figure presents the radial distribution function of the Ca2+ and Cl− ions in the EDTA and Ca2+ in water simulations, where it is apparent that the Ca2+ atoms are attached to the EDTA molecule. Table shows the values of the averages of the distance, minimum distance, and percentage dwell time of the counter ions (Cl− and Ca2+) included in the simulations. The average values and their standard deviations are the average over all the ions of the same type over all the trajectories from the three MD simulations performed here for this system.
Figure 1. Average of the radial distributions of the Ca2+ and Cl− ions over the three runs performed for the EDTA and Ca2+ system in water.
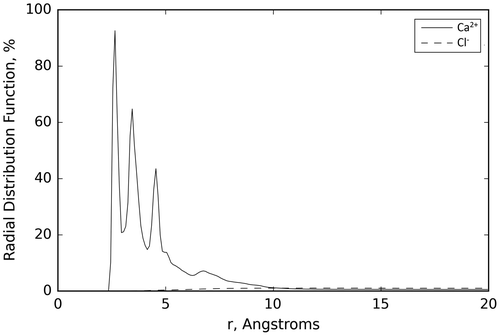
Table 1. Average distance and average minimum distance from the van der Waals surface of the EDTA molecule and average percentage dwell time of the counter ions (Cl− and Ca2+) included in this simulation
Both the figure and table present results that are indicative of the ability of EDTA to chelate Ca2+ in water. As it can be seen in Table , the average minimum distance, 2.86 Å, from the surface of EDTA for the Ca2+ ions shows that in all the simulations the Ca2+ ions have migrated, at least temporarily, from a distance of 8.00 Å to a distance within 3.00 Å of the surface of the EDTA molecule. The average distance of the Ca2+ ions to the surface of the EDTA molecule is comparably large, but this is largely due to the fact that EDTA has only four terminal COOH groups. This allows for only two Ca2+ ions to bind to EDTA. To ensure that the same concentration of Ca2+ ions was used for all the MD simulation studies in water, five Ca2+ ions were used in the EDTA and Ca2+ in water MD simulations, therefore not all the Ca2+ ions can be in close proximity to the EDTA molecule, resulting in a larger average distance and standard deviation. This is also apparent from Figure , where the Ca2+ ions show secondary peaks corresponding to second and third absorption layers. The other two Ca2+ ions are at an average distance of ~2.5 Å, which corresponds to a situation in which the ions are bound to EDTA. This is confirmed by the average percentage dwell time for Ca2+ ions, which shows that for most of the simulation, 80% of the time, at least two of the Ca2+ ions are interacting with the surface of EDTA. These simulations’ results agree with the experimental results showing the large affinity of EDTA to chelate Ca2+ (Amsterdam & Jamieson, Citation1974).
The small standard deviations observed for the average minimum distance indicate that all ions at a given time in the simulation visit the proximity of the EDTA molecule, while the large standard deviations observed both for the average distance and average percentage dwell time of Ca2+ are an indication that there is dynamic equilibrium in which preferentially these ions are close to the EDTA surface. The small deviation on the Cl− ion values indicates that these ions remain far from the EDTA surface; an expected result based on the EDTA and Cl− polarity.
3.2. MD simulation study of EDTA and Ca2+ in a buffer solution
Results from the MD simulation study of EDTA and Ca2+ in a buffer are presented in Table , which shows the average distance, minimum distance, and average percentage dwell time of all the counter ions (Cl−, Na+, and Ca2+) in the buffer solution, as well as their standard deviations of these values when averaged over all the trajectories in the three simulations performed here for this system.
Table 2. Average distance and average minimum distance from the van der Waals surface of the EDTA molecule and average percentage dwell time of the counter ions (Cl−, Na+, and Ca2+) included in this simulation
The average minimum distance of Ca2+ ions to the surface of the EDTA molecule is 2.54 Å, slightly smaller than the one observed in the water simulation and within 3.00 Å of the surface of the EDTA molecule. The average distance of the Ca2+ ions is 9.05 Å, also slightly smaller than in the water simulations. Its relatively large value can be explained by the same argument used for the simulations in water. The average dwell time for the Ca2+ ions also indicates that on average two Ca2+ ions are interacting with the surface of EDTA for a majority of the MD simulations. The results in the table show that the Na+ ions are much less likely to be close to the EDTA surface. It is not surprising that the minimum distance of Na+ (2.02 Å) is closer than that observed in the Ca2+, the Na+ ions are smaller and they only interact with one surface group on the EDTA molecule, whereas the Ca2+ ions commonly interact with two surface groups on the EDTA molecule. The average distance and the average percentage dwell time are considerably larger and smaller, respectively. These results show that while the Na+ is able to get in proximity of the EDTA, its interaction is less favorable than for Ca2+ as demonstrated by the larger average distance and smaller percentage dwell time. The corresponding radial distribution from this simulation (not shown) is similar to the one for the EDTA Ca2+ in water confirming the results reported in Table .
The results in the buffer solution are similar to those observed in the simulations of EDTA and Ca2+ in water. While it can be observed in the simulations that there is some competition for binding sites on EDTA between the six Na+ ions and two Ca2+ ions in the simulations, when comparing the results in Tables and it is apparent that this competition did not affect EDTA’s ability to chelate Ca2+. This indicates that the ability of EDTA to chelate Ca2+ is not affected by the presence of a competing ion (Na+) in the simulation environment. The results of the MD simulations are consistent with experimental results discussed above (Tomita et al., Citation1996; Vllasaliu et al., Citation2012).
The simulations results agree with the experimental results showing the ability of EDTA to chelate Ca2+ (Amsterdam & Jamieson, Citation1974) and validate our modeling approach to study chelation of G3.5 PAMAM dendrimers.
3.3. MD simulation study of G3.5 PAMAM dendrimer and Ca2+ in water
Results from the MD simulations of G3.5 PAMAM dendrimer and Ca2+ are presented in Figure and Table . Figure shows a three-dimensional depiction of the final recorded step of one of the simulations of G3.5 PAMAM dendrimer with Ca2+ in water, while Table shows the values for the average distance and minimum distance from the surface of the G3.5 PAMAM dendrimer molecule and average percentage dwell time of the Ca2+ ions, as well as their standard deviations calculated over all the trajectories in the three independent simulations performed for this system.
Figure 2. Three-dimensional representation of the final recorded step of one of the MD simulations of the G3.5 PAMAM dendrimer and Ca2+ in water.
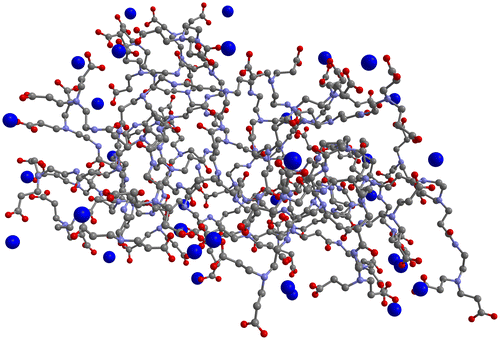
Table 3. Average distance and average minimum distance from the van der Waals surface of the G3.5 PAMAM molecule and average percentage dwell time of the counter ions (Ca2+) included in this simulation
Both the figure and table present results that clearly show the ability of G3.5 PAMAM dendrimer to chelate Ca2+. Figure shows that multiple Ca2+ ions are interacting with the surface of the G3.5 PAMAM dendrimer molecule in the final recorded step of one of the MD simulation runs. As can be seen in Table , the average minimum distance of Ca2+ from the surface of G3.5 PAMAM dendrimer is 2.40 Å, which is closer than the average minimum distance of Ca2+ observed in the EDTA simulations in water (2.86 Å). Also, the average distance of the Ca2+ ions to the surface of the G3.5 PAMAM dendrimer molecule, 4.13 Å, is much smaller than that observed with EDTA (10.34 Å) in water consistent with the fact that in this system all Ca2+ ions can be bound to the G3.5 PAMAM at the same time. The average percentage dwell time of 0.86 shows that the G3.5 PAMAM dendrimer is nearly always, almost 90% of the time, binding Ca2+ ions. Note that the decrease in the average distance and dwell time is also associated with the fact that the calcium ions in this simulation are in the correct stoichiometric ratio, such that all the ions can be binding to the G3.5 PAMAM dendrimer at the same time.
3.4. MD simulation study of G3.5 PAMAM dendrimer and Ca2+ in a buffer solution
Results from the MD simulation study of the G3.5 PAMAM dendrimer and Ca2+ in a buffer are presented in Table that shows the values for the average distance and minimum distance from the surface of the G3.5 PAMAM dendrimer molecule and the percentage dwell time of all the counter ions (Na+ and Ca2+) present in the buffer solution, as well as their standard deviations calculated over all trajectories from the three independent MD simulations performed in this system.
Table 4. Average distance and average minimum distance from the van der Waals surface of the G3.5 PAMAM molecule and average percentage dwell time of the counter ions (Na+ and Ca2+) included in this simulation
Figure shows the average of the radial distribution functions of all the ions considered here over the three runs performed for the G3.5 PAMAM dendrimer and Ca2+ system in water and the G3.5 PAMAM dendrimer in a buffer solution. It is apparent from the figure that (i) all the Ca2+ ions are clearly attached to the G3.5 PAMAM dendrimer and (ii) that the introduction of potentially competitive Na+ ions does not change the chelation ability of Ca2+ ions by the G3.5 PAMAM dendrimers. The ability of Na+ to bind to the G3.5 PAMAM was independently verified by simulations without Ca2+ ions (supplementary material Table S5 and Figure S6).
Figure 3. Average of the radial distribution functions of the Ca2+ ions over the three runs performed for the G3.5 PAMAM dendrimer and Ca2+ system in water (top) and the Ca2+ and Na+ ions over the three runs performed for the G3.5 PAMAM dendrimer in a buffer solution (bottom).
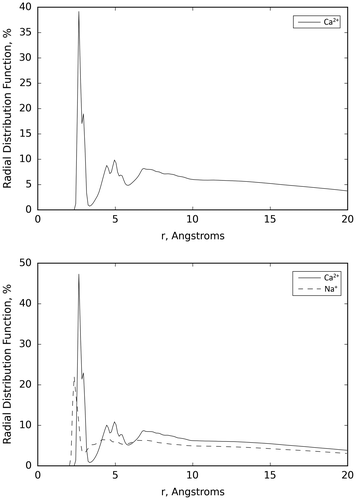
These results show that the results obtained in water can also be expected in a buffer solution that mimics a situation closer to the intestinal lumen milieu and that the preferential binding is not sensitive to the details of the solution used in the simulations. Moreover, the preferential binding of Ca2+ is highlighted by the difference between the average distance of each ion to the surface of the G3.5 PAMAM. The average distance of the Ca2+ ions to the surface of the G3.5 PAMAM dendrimer molecule, 3.55 Å, is much smaller than that observed with the Na+ ions, 7.50 Å. This indicates that the Ca2+ ions appear to be more frequently bound to the G3.5 PAMAM dendrimer than the Na+ ions. The average minimum distance of Ca2+ from the surface of G3.5 PAMAM dendrimer of 2.40 Å is consistent with what was observed in the G3.5 PAMAM dendrimer and Ca2+ simulation in water.
The average minimum distance of Na+ (2.02 Å) is shorter than that observed in the Ca2+, in agreement with the results discussed above for the simulation of EDTA in buffer. Results of the average percentage dwell time of the Na+ and Ca2+ ions show a relatively large difference, 0.60 for Na+ and 0.91 for Ca2+ ions, which is similar to that observed in the simulation in water. This indicates that the Ca2+ ions appear to be more frequently bound to the G3.5 PAMAM dendrimer than the Na+ ions. Results show that Ca2+ is an effective chelation agent for G3.5 PAMAM dendrimer, indicating that under the simulation conditions the G3.5 dendrimer exhibits high chelating efficiency of Ca2+ in both water and in a buffer solution.
3.5. Apt computational environment
The use of AMBER in the Apt HPC environment has been extremely successful. During the course of these studies we were able to secure all the needed resources for the project, and because Apt is able to mirror a standard HPC environment, the migration to this cloud-based computing environment has been straightforward. Moreover, the overhead for using the Apt environment has been minimal. The only overhead incurred when using Apt is the instantiation and de-provisioning of the infrastructure, which is typically less than 15 min, a small fraction of a typical AMBER simulation.
4. Conclusions
Using MD simulations, validated by the agreement with existing experimental results in EDTA, we have shown that in MD simulations G3.5 PAMAM dendrimers exhibit a high chelating efficiency of Ca2+ comparable to the EDTA efficiency and therefore they may be capable of using this mechanism to open the tight junctions in the intestines. We have demonstrated that computationally intensive applications of interest in drug delivery research can effectively use cloud computing environments like Apt.
Funding
This work was supported by National Institutes of Health [grant number T15LM007124], [grant number 1ULTR001067], [grant number ES024681]; National Science Foundation [grant number CNS-1338155].
Supplementary_material.docx
Download MS Word (2.4 MB)Acknowledgments
The authors are grateful to University of Utah Center for High Performance Computing for providing computing resources.
Additional information
Notes on contributors
David E. Jones
David E. Jones received his PhD in Biomedical Informatics working under the supervision of professors Hamidreza Ghandehari and Julio C. Facelli. He was part of the program in Informatics of Nanoscale Drug Delivery Systems for Cancer Treatment. This program’s research focus is in areas of nanomedicine related to the use of nanoparticles as delivery vectors for pharmaceutics, diagnostic devices, and tissue replacement materials. In this project, we develop and adapt text mining, data extraction, data mining, chemometrics, and molecular modeling techniques to address important questions in nanomedicine with emphasis on drug delivery systems for cancer treatment. The ultimate goal of the project is to develop in silico environments to accelerate the development of nanoscale drug delivery systems for cancer treatment.
Related Research Data
References
- Amsterdam, A., & Jamieson, J. D. (1974). Studies on dispersed pancreatic exocrine cells: I. Dissociation technique and morphologic characteristics of separated cells. The Journal of Cell Biology, 63, 1037–1056.10.1083/jcb.63.3.1037
- Astachov, V., Garzoni, M., Danani, A., Choy, K. L., Pavan, G. M., & Fahmi, A. (2016). In situ functionalization of self-assembled dendrimer nanofibers with cadmium sulfide quantum dots through simple ionic-substitution. New Journal of Chemistry, 40, 6325–6331.
- Avila-Salas, F., Sandoval, C., Caballero, J., Guiñez-Molinos, S., Santos, L. S., Cachau, R. E., & González-Nilo, F. D. (2012). Study of interaction energies between the PAMAM dendrimer and nonsteroidal anti-inflammatory drug using a distributed computational strategy and experimental analysis by ESI-MS/MS. The Journal of Physical Chemistry B, 116, 2031–2039.10.1021/jp2069122
- Barata, T., Teo, I., Lalwani, S., Simanek, E., Zloh, M., & Shaunak, S. (2011). Computational design principles for bioactive dendrimer based constructs as antagonists of the TLR4-MD-2-LPS complex. Biomaterials, 32, 8702–8711.10.1016/j.biomaterials.2011.07.085
- Cao, J., Zhang, H., Wang, Y., Yang, J., & Jiang, F. (2013). Investigation on the interaction behavior between curcumin and PAMAM dendrimer by spectral and docking studies. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, 108, 251–255.10.1016/j.saa.2013.02.003
- Case, D. A., Babin, V., Berryman, J. T., Betz, R. M., Cai, Q., Cerutti, D. S., … Kollman, P. A. (2014). AMBER 14. San Francisco, CA: University of California.
- Castriciano, M. A., Romeo, A., Angelini, N., Micali, N., Guccione, S., & Scolaro, L. M. (2011). Spectroscopic investigation and molecular modeling on porphyrin/PAMAM supramolecular adduct. Photochemistry and Photobiology, 87, 292–301.10.1111/php.2011.87.issue-2
- ChemAxon. (2013). Marvin 5.12.4. Cambridge, MA: ChemAxon.
- ChemAxon., Berry, I., Ruyts, B. (2012). Future-proofing cheminformatics platforms (pp. 1–16). Retrieved October 31, 2013, from http://www.chemaxon.com/wp-content/uploads/2012/04/Future_proofing_cheminformatics_platforms.pdf
- Garzoni, M., Cheval, N., Fahmi, A., Danani, A., & Pavan, G. M. (2012). Ion-selective controlled assembly of dendrimer-based functional nanofibers and their ionic-competitive disassembly. Journal of the American Chemical Society, 134, 3349–3357.10.1021/ja206611s
- Hubbard, D., Ghandehari, H., & Brayden, D. J. (2014). Transepithelial transport of PAMAM dendrimers across isolated rat jejunal mucosae in ussing chambers. Biomacromolecules, 15, 2889–2895.10.1021/bm5004465
- Hubbard, D., Enda, M., Bond, T., Moghaddam, S. P., Conarton, J., Scaife, C., ... Ghandehari, H. (2015). Transepithelial transport of PAMAM dendrimers across isolated human intestinal tissue. Molecular Pharmaceutics, 12, 4099–4107.10.1021/acs.molpharmaceut.5b00541
- Ivanov, A. A., & Jacobson, K. A. (2008). Molecular modeling of a PAMAM-CGS21680 dendrimer bound to an A2A adenosine receptor homodimer. Bioorganic & Medicinal Chemistry Letters, 18, 4312–4315.10.1016/j.bmcl.2008.06.087
- Jensen, L. B., Mortensen, K., Pavan, G. M., Kasimova, M. R., Jensen, D. K., Gadzhyeva, V., ... Foged, C. (2010). Molecular characterization of the interaction between siRNA and PAMAM G7 dendrimers by SAXS, ITC, and molecular dynamics simulations. Biomacromolecules, 11, 3571–3577.10.1021/bm101033g
- Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W., & Klein, M. L. (1983). Comparison of simple potential functions for simulating liquid water. The Journal of Chemical Physics, 79, 926–935.10.1063/1.445869
- Karatasos, K., Posocco, P., Laurini, E., & Pricl, S. (2012). Poly(amidoamine)-based dendrimer/siRNA complexation studied by computer simulations: Effects of pH and generation on dendrimer structure and siRNA binding. Macromolecular Bioscience, 12, 225–240.10.1002/mabi.v12.2
- Lee, I., Majoros, I. J., Williams, C. R., Athey, B. D., & Baker, J. R. (2009). Interactive design strategy for a multi-functional PAMAM dendrimer-based nano-therapeutic using computational models and experimental analysis. Journal of Computational and Theoretical Nanoscience, 6, 54–60.10.1166/jctn.2009.1006
- Noach, A. B., Kurosaki, Y., Blom-Roosemalen, M. C., & de Boer, A. G. (1993). Cell-polarity dependent effect of chelation on the paracellular permeability of confluent caco-2 cell monolayers. International Journal of Pharmaceutics., 90, 229–237.10.1016/0378-5173(93)90195-L
- Roe, D. R., & Cheatham, 3rd, T. E. (2013). PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. Journal of Chemical Theory and Computation, 9, 3084–3095.10.1021/ct400341p
- Shinoda, W., DeVane, R., & Klein, M. L. (2012). Computer simulation studies of self-assembling macromolecules. Current Opinion in Structural Biology, 22, 175–186.10.1016/j.sbi.2012.01.011
- Tomita, M., Hayashi, M., & Awazu, S. (1996). Absorption-enhancing mechanism of EDTA, caprate, and decanoylcarnitine in Caco-2 cells. Journal of Pharmaceutical Sciences, 85, 608–611.10.1021/js9504604
- Vasumathi, V., & Maiti, P. K. (2010). Complexation of siRNA with dendrimer: A molecular modeling approach. Macromolecules, 43, 8264–8274.10.1021/ma1012495
- Vllasaliu, D., Shubber, S., Garnett, M., Alexander, C., Eaton, M., & Stolnik, S. (2012). Evaluation of calcium depletion as a strategy for enhancement of mucosal absorption of macromolecules. Biochemical and Biophysical Research Communications, 418, 128–133.10.1016/j.bbrc.2011.12.146
- Wang, J., Wang, W., Kollman, P. A., & Case, D. A. (2006). Automatic atom type and bond type perception in molecular mechanical calculations. Journal of Molecular Graphics and Modelling., 25, 247–260.10.1016/j.jmgm.2005.12.005
- Wang, B., Navath, R. S., Menjoge, A. R., Balakrishnan, B., Bellair, R., Dai, H., ... Kannan, R. M. (2010). Inhibition of bacterial growth and intramniotic infection in a guinea pig model of chorioamnionitis using PAMAM dendrimers. International Journal of Pharmaceutics, 395, 298–308.10.1016/j.ijpharm.2010.05.030
- Xiang, T. X., & Anderson, B. D. (2006). Liposomal drug transport: A molecular perspective from molecular dynamics simulations in lipid bilayers. Advanced Drug Delivery Reviews, 58, 1357–1378.10.1016/j.addr.2006.09.002