
Abstract
Liposome electrokinetic chromatography and micellar electrokinetic chromatography were used for studying the lipophilic properties of natural antioxidants, specifically phenolic acids and flavonoids. The employed negatively charged liposomes were composed of mixtures of 1-palmitoyl-2-oleyl-sn-glycerophosphatidylcholine and 1-palmitoyl-2-oleoyl-sn-phosphatidylserine or 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylglycerol. In micellar electrokinetic chromatography, sodium dodecyl sulphate micelles were used as the pseudostationary phase. The retention factors of the studied compounds were determined at pH 7.4. The corresponding distribution constants were calculated from the experimentally determined retention factors and the phase ratios of the liposome dispersions and the micellar system. The distribution constants between the aqueous phase and the liposomes or micelles were compared with octanol/water partition or distribution constants of the studied compounds, which were predicted using the ACD/Labs Percepta Platform—PhysChem Module. Our results indicate that the correlations between the distribution constants of the two tested liposome systems were much stronger than those between the liposome/micellar systems. The correlations between the n-octanol phase and the liposome phases were similar to that between n-octanol and the micellar phase. Our data shows that electrokinetic chromatography is an efficient method for determining partition coefficients of compounds, but the type of pseudostationary phase has a clear impact on the values.
Public Interest Statement
Lipophilicity is a property of a compound, which is related to its solubility between water and an organic (lipid) phase. Lipophilic compounds often display high volumes of distribution and are therefore distributed amongst many different tissues and organs. The most common surrogate for lipophilicity is the octanol/water partition coefficient. Here, we used micellar electrokinetic chromatography (MEKC) and liposome electrokinetic chromatography (LEKC) for studying the lipophilic properties of natural antioxidants. By measuring the interactions between the compounds and the micelles or liposomes, we were able to determine the corresponding distribution constants of the compounds. Such values are of great importance for people in the field of pharma and medical science, because compounds’ lipophilicity is one of the most important properties determining the distribution of compounds in the body.
Competing Interests
The authors declare no competing interest.
1. Introduction
Antioxidants are secondary metabolites of plants, which can protect organisms from the effect of free radicals. Antioxidants are able to inhibit oxidative damages and to improve the immune function of organisms (Citation1). In vivo effects of these compounds are dependent on their lipophilicity and hydrophilicity, which govern membrane and protein interactions. In general, the hydrophobicity or lipophilicity of compounds can be expressed by the distribution constant between a polar and a non-polar phase (Citation2). The octanol/water partition coefficient and its logarithm, log Po/w, are commonly used for the description of the hydrophobicity or lipophilicity of compounds. These values are also good descriptors for the characterisation of the relationship between the structure and biological, pharmacological and ecological effects of the compounds (Citation3). However, for charged (ionised) compounds, it is important to consider the distribution constant between n-octanol and a buffer with the target pH value (Citation4). To get a better understanding of the distribution of compounds between lipophilic and hydrophilic phases, micelle or liposome/aqueous partition coefficients, which are dependent on van der Waals and hydrogen donor/acceptor interactions between the compounds and the lipophilic membrane, can be determined (Citation5).
In this work, we used micellar electrokinetic chromatography (MEKC) and liposome electrokinetic chromatography (LEKC) for the separation of some antioxidants commonly present in plant material. MEKC has been widely employed for the separation of synthetic and natural drugs (Citation6, 7), proteins and peptides (Citation8, 9), oligonucleotides (Citation10), carbohydrates (Citation11, 12) and for the separation of natural antioxidants (Citation13–16). The advances in developing fundamental aspects and applications of MEKC have been summarised, e.g. in “Silva 2011 (Citation17)”. LEKC has been known since the mid-90s, when it was used for characterisation of interactions between lipid bilayers and peptides, or drugs (Citation18). Later on, Roberts et al. (Citation19) studied the behaviour of liposomes in capillary electrophoresis. Since then, several papers on LEKC have been published (Citation20–22). Even though liposomes are rather crude biomimetic models of biological cell membranes, data has shown that LEKC can be used for predicting the behaviour of compounds in living systems (Citation5, 23, 24).
The anti-atherosclerotic, anti-inflammatory, antitumor, antibacterial, antiviral and antitrombogenic effects of natural antioxidants are described in the literature (Citation25–27). Polyphenols, including flavonoids, are the most common natural antioxidants in the human diet. Flavonoids are divided into six groups—flavones, flavonoles, flavanoles, flavanones, isoflavones and anthocyanines. The differences between the groups are given by the varied degree of hydroxylation, methoxylation or glycosylation. Coumarins and phenolic acids represent the simple phenols. The phenolic acids can further be divided into two groups based on the structure of cinnamic acid or benzoic acid. In vivo effects of these compounds are highly dependent on their lipophilicity and hydrophilicity, and therefore, in this study, micelle/aqueous phase and liposome/aqueous phase distribution constants of antioxidants were determined by MEKC and LEKC, respectively. Both the sodium dodecyl sulphate (SDS) micelles and the employed liposomes were negatively charged. The liposomes comprised of 1-palmitoyl-2-oleyl-sn-glycero-3-phosphocholine (POPC) and 1-palmitoyl-2-oleyl-sn-glycero-3-phospho-L-serine (POPS) or 1-palmitoyl-2-oleyl-sn-glycero-3-phospho-(1′-rac-glycerol) (POPG). All lipids chosen have transition temperatures well below room temperature, meaning that the liposomes used were in the disordered liquid crystalline phase throughout the study. To our knowledge, this is the first time a direct EKC comparison of LEKC and MEKC, using the same volume of the pseudostationary phase (similar phase ratio), has been done.
2. Materials and methods
2.1. Materials and reagents
1-Palmitoyl-2-oleyl-sn-glycero-3-phosphocholine (POPC), 1-palmitoyl-2-oleyl-sn-glycero-3-fosfo-L-serine (POPS) and 1-palmitoyl-2-oleyl-sn-glycero-3-phosphoglycerol (POPG) were purchased from Avanti Polar Lipids (Alabaster, AL, USA). The purity of all phospholipids were >99% according to Avanti Polar Lipids home page (https://avantilipids.com/). The phase transition temperatures of POPC, POPS and POPG are −2, 14 and −2°C, respectively. The structures of the phospholipids are shown in the supporting information (SI), Figure S2. SDS, the alkylbenzoates and the standards of the antioxidants (Figure S3) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Sodium hydrogen phosphate and thiourea were from Sigma (Darmstadt, Germany) and sodium dihydrogen phosphate monohydrate and HPLC-grade methanol were purchased from Mallinckrodt Baker (Deventer, The Netherlands). Methanol was purchased from VWR International (Espoo, Finland) and sodium hydroxide from FF-Chemicals (Yli-Ii, Finland). All compounds were used as received.
2.2. Capillary electrophoresis
An Agilent 7100 (Agilent, Palo Alto, CA, USA) capillary electrophoresis instrument equipped with a diode array detector was used for the LEKC and for the MEKC studies. Uncoated fused-silica capillaries with 50 μm I.D. (360 μm O.D.) and a total length of 36.5 cm (effective length of 28 cm) were used throughout the study. When a new capillary was taken into use, it was preconditioned by rinsing (pressure of 940 mbar) for 15 min with NaOH (0.1 M), for 15 min with Milli-Q water and for 15 min (MEKC conditions) or 5 min (LEKC conditions) with the background electrolyte (BGE) solution before the first run. The sample was injected hydrodynamically by applying 50 mbar pressure for 5 s on the inlet sample vial. The applied voltage was 20 kV, the temperature was 25°C and the detection wavelength was set at 214 and 254 nm. All capillary zone electrophoresis (CZE) and MEKC experiments were repeated five times. For LEKC experiments, the total volume of the BGE was only 225 μL (due to the high cost of phospholipids), and therefore, the LEKC experiments were repeated only three times.
2.3. Preparation of buffer, liposomes, micelles and antioxidant samples
Phosphate buffer (pH 7.4; ionic strength of 20 mM) was prepared by mixing appropriate amounts of solutions of sodium hydrogen phosphate and sodium dihydrogen phosphate. The total concentration of phosphate anion formed was 8.2 mM, and the concentration of sodium cations was 14.1 mM. The phosphate buffer was filtered through a 0.45 μm membrane filter before use. The BGE for MEKC contained SDS (5.2 mM) in phosphate buffer (pH 7.4; ionic strength of 20 mM). The BGE for LEKC was prepared from stock solutions of POPC (20 mM), POPS (12.7 mM) and POPG (10 mM) in chloroform (stored in a freezer) and phosphate buffer (pH 7.4; ionic strength of 20 mM). Appropriate amounts of the stock solutions of the phospholipids were mixed and evaporated to dryness under a gentle stream of air. Traces of chloroform were removed by evacuation under 8-100 mbar pressure for 20 h. The lipid film was hydrated with phosphate buffer and shaken at 1,000 rpm for 60 min at 60°C. The liposome dispersion was extruded 19 times through a Nucleopore Track-Etch membrane (Whatman, Maidstone, UK) with a pore size of 100 nm. The total concentrations of the liposome dispersions, i.e. POPC/POPS (80:20, mol%) and POPC/POPG (80:20, mol%), were 4 mM, which were further diluted to 0.5 mM with phosphate buffer for LEKC analyses.
The concentration of the stock solution of each antioxidant compound was 1 g/L in methanol. The concentration of the antioxidants injected for analyses was 25 mg/L in Milli-Q water, except for morin, biochanin A, esculin, isoquercitrin, luteolin and myricetin, which were of 50 mg/L in water/methanol 3:1 (v/v). Thiourea (0.5 mM solution in phosphate buffer) was used as a marker of the electroosmotic flow (EOF).
2.3.1. Calculation of retention factors and determination of distribution constants
A description of the determination of the retention factors is given in the supporting information (SI). The effective mobilities of the micelles and the liposomes were determined using an iterative procedure, utilising a series of alkylbenzoate homologues (methylbenzoate to hexylbenzoate) (Citation28, 29). The scheme of the iterative procedure is shown in the supporting information (Figure S1). The distribution constants of the compounds were calculated from the retention factors. The equations and details about the calculations are given in the supporting information.
3. Results and discussion
3.1. Determination of the electrophoretic mobility of liposomes and micelles
Determination of the electrophoretic mobilities of the liposomes and micelles is necessary for calculating the retention factors of the compounds in LEKC and MEKC. The iterative procedure using a homologous series of alkylbenzoates (C1–C6) was used for the determination of the electrophoretic mobility of the liposomes comprising POPC/POPS or POPC/POPG and for the SDS micelles. The mobility of 80:20 mol% POPC/POPS liposomes with a concentration of 0.5 mM in phosphate buffer (pH 7.4, 20 mM ionic strength) was −4.18 × 10−8 m2 V−1 s−1. The electrophoretic mobility of 80:20 mol% POPC/POPG liposomes was −4.33 × 10−8 m2 V−1 s−1. These values are rather similar to the values previously obtained for liposomes and lipid emulsions, i.e. Intralipid® and ClinOleic® (Citation24, 30–32).
The electrophoretic mobility of the SDS micelles was −4.57 × 10−8 m2 V−1 s−1, which is in good agreement with previously published values (Citation33, 34). Both liposomes and SDS micelles have an overall negative surface charge, but the smaller size of SDS micelles in comparison with the liposomes explains the higher mobility of the SDS micelles.
3.2. Determination of the distribution constants of natural antioxidants
The distribution constants between the phosphate buffer (pH 7.4, ionic strength of 20 mM) and the liposomes, i.e. 80:20 mol% POPC/POPS or 80:20 mol% POPS/POPG, or the SDS micelles were determined from the effective electrophoretic mobilities of the flavonoids and phenolic acids measured under CZE, LEKC and MEKC conditions. Typical electropherograms under CE (CZE) and LEKC conditions are shown in Figure S4. The obtained values were compared with the values of octanol/aqueous distribution constants. The values of log DpH 7.4 were used for ionised compounds and log Po/w values for neutral compounds (flavone, (+)-catechin). Log DpH 7.4 and log Po/w values were predicted using the ACD/Labs Percepta Platform—PhysChem Module and the pKa values were predicted using Advanced Chemistry Development (ACD/Labs) Software V11.02 (©1994–2015 ACD/Labs). The values are listed in Table .
Table 1. Values of pKa, log DpH 7.4, log Po/w and experimentally determined distribution constants (log KD) of phenolic acids and flavonoids
3.3. Comparison of the distribution constants with log DpH 7.4 or log Po/w values
The degree of ion dissociation and the experimentally determined distribution constants for the phenolic acids and flavonoids using MEKC and LEKC are also listed in Table . The correlation between the obtained log KD (POPC/POPS) values and the predicted log DpH 7.4/log Po/w values is shown in Figure (A). There are two groups of studied compounds presented in the graph; group I represents the phenolic acids, the flavonoid glycosides (esculin, rutin and isoquercitrin) and 4-hydroxycoumarin; group II represents the rest of the compounds, except for hesperidin, naringin and 4-hydroxybenzoic acid. Hesperidin and naringin belong to the group of flavonoid glycosides (flavanones), and they did not interact with the POPC/POPS liposomes under the selected experimental conditions (the retention factors were equal to zero). The reason for this could be due to the very low hydrophobicity of the compounds, indicated by their log DpH 7.4 values of −0.26 for hesperidin and −0.05 for naringin. This polar character in combination with their very bulky structures can at least partly explain the lack of interactions with the POPC/POPS liposomes.
Figure 1. (A) Correlation between log KD (POPC/POPS) values and log DpH 7.4/log Po/w and (B) correlation between log KD (POPC/POPG) values and log DpH 7.4/log Po/w. Compounds: (1) gallic acid; (3) protocatechuic acid; (4) salicylic acid; (5) syringic acid; (6) vanillic acid; (7) caffeic acid; (8) chlorogenic acid; (9) ferulic acid; (10) p-coumaric acid; (11) sinapic acid; (12) (+)-catechin; (13) (−)-epicatechin; (14) 7-hydroxyflavon; (15) biochanin A; (16) flavone; (17) isoquercitrin; (18) luteolin; (19) rutin; (20) kaempferol; (21) morin; (22) myricetin; (23) quercetin; (24) hesperetin; (26) naringenin; (28) 4-hydroxycoumarin; (29) 6,7-dihydroxycoumarin; (30) esculin. Data was evaluated using program Statistica 12 and graphical program Origin 9. Running conditions: uncoated fused-silica capillary (50 μm I.D./360 μm O.D, ltot = 36.5 cm, ldet = 28.0 cm); BGE: 0.5 mM 80/20 mol% POPC/POPS (A) or 0.5 mM 80/20 mol% POPC/POPG (B) in phosphate buffer at pH 7.4 (I = 20 mM); separation voltage + 20 kV; capillary cassette temperature 25°C; sample injection 50 mbar/5 s; UV detection at 214 and 254 nm; 0.5 mM thiourea was used as an EOF mobility marker.
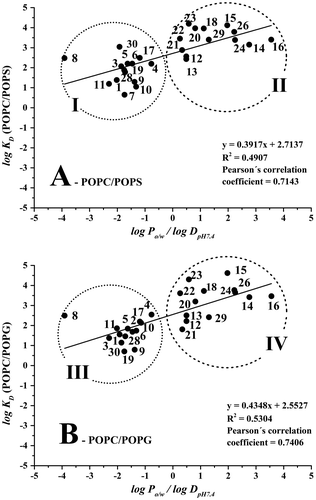
In the following step, the interactions between the compounds and 80:20 mol% POPC/POPG liposomes were studied. The obtained log KD (POPC/POPG) values were compared with the predicted octanol/aqueous distribution constants. The correlation between the liposome (POPC/POPG)/aqueous and octanol/aqueous distribution constants is shown in Figure (B). Similarly, to the POPC/POPS liposomes, the compounds are divided into two groups. Phenolic acids (excepted for caffeic acid), glycosylated flavonoids and 4-hydroxycoumarin are clustered in group III, whereas the aglycones of flavonoids (except for hesperidin and naringin) are clustered in group IV. The groups III and IV generally correspond to groups I and II presented in Figure (A). Due to negligible interactions with the liposomes, the retention factors of caffeic acid, hesperidin and naringin were equal to zero.
The positive slope of the regression line in Figure indicates that the antioxidants prefer to interact with the hydrophobic phospholipid membrane. The polar compounds (group I in Figure (A) and group III in Figure (B)) are only weakly retained in both liposome systems, with the slope of the correlation line being higher for the POPC/POPG liposomes. According to the data presented in Table , higher log KD values were observed for some phenolic acids (i.e. gallic acid, 4-hydroxybenzoic acid, p-coumaric acid and sinapic acid) when comparing POPC/POPG liposomes with POPC/POPS liposomes. This clearly indicates selective interactions between some of the compounds and the polar head groups of the lipids. Even though both phospholipids, i.e. POPG and POPS, have an overall negative surface charge, the polar parts of the molecules are dissimilar. POPS has two negatively charged functional groups and one neutralising positively charged group, whereas POPG only contains one negatively charged phosphate group. The 1,2-diol presented in POPG can also specifically interact with the aforementioned compounds.
3.4. Correlation of experimental and predicted distribution constants
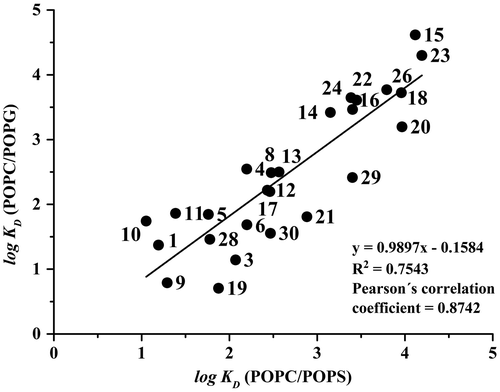
For detailed characterisation of the correlation between the log KD data, the Pearson’s correlation coefficient, r, was used. Pearson’s correlation coefficient is one of the most important and most commonly used parameters for measuring the strength of a correlation between two continuous random variables. Pearson’s correlation coefficient is expected from N pairs of values—the correlation pairs of the measured N individuals randomly selected from the population. Pearson’s correlation coefficient determined for the POPC/POPS liposomes vs log Po/w/log DpH7.4 was 0.714 (R2 = 0.491) and for the POPC/POPG liposomes vs log Po/w/log DpH7.4 the positive correlation was 0.740 (R2 = 0.530). The values of the Pearson’s correlation coefficients are in the range of 0.7–0.8, which is typical for strong correlation between studied variables. The Pearson’s correlation coefficient was even higher (r = 0.874, N = 25, p < 0.05) for the correlation between log KD (POPC/POPS) and log KD (POPC/POPG), shown in Figure . This demonstrates that the positive correlation between the two liposome systems is stronger than between the log KD values obtained using the liposome systems and the predicted log Po/w/log DpH 7.4 values.
Figure 2. Correlation between log KD (POPC/POPG) values and log KD (POPC/POPS) values. The numbering of the compounds and the running conditions are the same as in Figure .
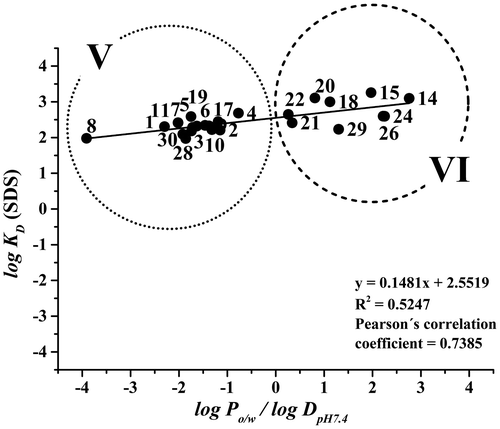
The pseudostationary phase in MEKC comprised 5.2 mM of SDS. The cmc of SDS was determined using a CE instrument for measuring the current as a function of increasing SDS concentration in a capillary filled with a BGE containing SDS (Citation35). The cmc value of SDS in phosphate buffer (pH 7.4, I = 20 mM) was 4.1 ± 0.2 mM. This means that the BGE employed in this work (comprising 5.2 mM SDS), contained both SDS micelles (with the micelle concentration 1.1 mM) and free SDS unimers. The concentration of the pseudostationary phase was selected to provide the same phase ratio in the LEKC and MEKC analyses. Interactions between (+)-catechin, (−)-epicatechin, flavone, quercitrin, hesperidin or naringin and SDS micelles were not observed in the MEKC analyses. The correlation between the logarithms of micelle/aqueous phase distribution constants and the log Po/w/log DpH 7.4 values is shown in Figure . The corresponding data is presented in Table . Group V is formed by the phenolic acids and flavonoid glycosides, and group VI represents the flavonoids. In comparison with the LEKC systems, the positive slope of the correlation line is lower, indicating that the less polar compounds interact more strongly with the phospholipid membrane than with the SDS micelles. The correlation between log KD (SDS) and log Po/w/log DpH 7.4, represented by Pearson’s correlation coefficient (r = 0.739, N = 24, p < 0.05), is however similar to the values obtained for POPC/POPS and POPC/POPG liposome dispersions.
Figure 3. Correlations between log KD (SDS) values and log Po/w/log DpH 7.4 values. The numbering of the compounds is the same as in Figure 1. Running conditions: uncoated fused-silica capillary (50 μm I.D./360 μm O.D., ltot = 36.5 cm, ldet = 28.0 cm); BGE: 5.2 mM SDS in phosphate buffer at pH 7.4 (I = 20 mM); separation voltage +20 kV; capillary cassette temperature 25°C; sample injection 50 mbar/5 s; UV detection at 214 and 254 nm; 0.5 mM thiourea was used as an EOF mobility marker.
3.5. Comparison of the distribution constants in LEKC and MEKC
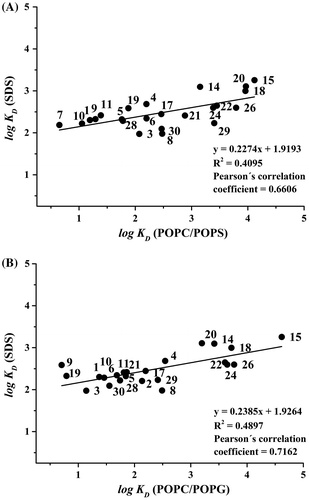
In order to compare the LEKC and MEKC separation systems and for characterisation of the distribution equilibria, the correlations between the constants determined for the SDS micelles and for the liposomes were constructed. The correlation between log KD (SDS) and log KD (POPC/POPS) is presented in Figure (A), and the correlation between log KD (SDS) and log KD (POPC/POPG) is shown in Figure (B). Both systems provided similar positive slopes of regression lines with slightly better correlation of SDS with POPC/POPG, indicated by a slightly higher Pearson’s correlation coefficient (0.716 for SDS-POPC/POPG compared to 0.661 for SDS-POPC/POPS). Micelles of SDS are negatively charged as well as the liposomes of POPC/POPG and POPC/POPS. However, at this stage, we are not able to comment on the reason why the Pearson’s correlation coefficient between SDS-POPC/POPG is higher than between SDS-POPC/POPS. The primary amino group presented in POPS could make a difference. The significant correlation of distribution constants by MEKC and LEKC suggests that the partition equilibria in SDS micelles can be used as a rough model for characterising the interaction of compounds with phospholipid membranes.
Figure 4. Correlations between (A) log KD (SDS) values and log KD (POPC/POPS); and (B) log KD (SDS) and log KD (POPC/POPG). The numbering of the compounds is the same as in Figure . Running conditions were as in Figures and .
4. Conclusions
In this work, liposome/aqueous and micellar/aqueous phase distribution constants of natural antioxidants were determined using LEKC and MEKC. Liposomes comprising 80:20 mol% POPC/POPS or 80:20 mol% POPC/POPG were used as the pseudostationary phase in LEKC, and SDS micelles were used in MEKC. In order to compare the distribution constants obtained by LEKC and MEKC, pseudostationary phases of similar phase ratios were utilised. The distribution constants were calculated from the phase ratio and the retention factors, and the KD values were compared with predicted values of octanol/aqueous phase distribution constants of the selected antioxidants. The results show that the distribution constants of phenolic acids and flavonoid glycosides are, in general, lower than the distribution constants of the flavonoids. Obviously, the type of polar head groups in the phospholipids or surfactants has a considerable effect on the distribution constants of the phenolic acids and flavonoids. The data indicated a stronger positive correlation between the two studied liposome systems than between the liposome/SDS systems.
Funding
This work was supported by the Academy of Finland [grant number 266342 (SW)] and from the Magnus Ehrnrooth Foundation [grant number 4703943 (SW)].
Supplementary material
Supplementary material for this article can be accessed here https://doi.org/10.1080/23312009.2017.1385173.
OACH_A_1385173_Supplementary_Material.docx
Download MS Word (935.5 KB)Additional information
Notes on contributors
Jana Váňová
Susanne K. Wiedmer is university lecturer at the Department of Chemistry at the University of Helsinki (Finland). Susanne Wiedmer’s group has expertise in chromatographic and capillary electromigration (CE) techniques, field flow fractionation (AF4) and in biosensing methods. Much focus has been on liposomes and on liposome-analyte interactions using various modes of CE, on studying interactions between biomembrane-imitating surfaces and analytes (by CE, nanoplasmonic sensing and quartz crystal microbalance), as well as on characterising lipid vesicles and particles (by CE and AF4). Recently, emphasis has been on the determination of distribution constants of analytes using liposomes of synthetic lipids or lipids extracted from biological samples. Ionic liquids have been another research target in the group, and focus has been on the determination of the toxicity of novel synthesised ionic liquids. Liposomes, cells and zebrafish models have been used for that purpose.
Related Research Data
References
- Jáč, P.; Polášek, M.; Pospíšilová, M. Recent Trends in the Determination of Polyphenols by Electromigration Methods. J. Pharm. Biomed. Anal. 2006, 40, 805–814.
- Rothwell, J.A.; Day, A.J.; Morgan, M.R.A. Experimental Determination of Octanol-Water Partition Coefficients of Quercetin and Related Flavonoids. J. Agric. Food Chem. 2005, 53, 4355–4360.10.1021/jf0483669
- Berthod, A.; Carda-Broch, S. Determination of Liquid-Liquid Partition Coefficients by Separation Methods. J. Chromatogr. A 2004, 1037, 3–14.10.1016/j.chroma.2004.01.001
- Kah, M.; Brown, C.D. LogD: Lipophilicity for Ionisable Compounds. Chemosphere 2008, 72, 1401–1408.10.1016/j.chemosphere.2008.04.074
- Wiedmer, S.K.; Kulovesi, P.; Riekkola, M.-L. Liposome Electrokinetic Capillary Chromatography in the Study of Analyte-Phospholipid Membrane Interactions. Application to Pesticides and Related Compounds. J. Sep. Sci. 2008, 31, 2714–2721.10.1002/jssc.v31:14
- Gong, F.; Yang, H.; Sun, W.; Cao, J.; Liu, W. Development and Validation of a Micellar Electrokinetic Capillary Chromatography Method for the Determination of Goserelin and Related Substances. Electrophoresis 2016, 37, 623–629.10.1002/elps.201500328
- Theurillat, R.; Sendi, P.; Thormann, W. An MEKC Assay for the Therapeutic Drug Monitoring of Cefepime. J. Sep. Sci. 2013, 36, 2915–2921.10.1002/jssc.201300402
- Glavač, N.K.; Injac, R.; Kreft, S. Optimization and Validation of a Capillary MEKC Method for Determination of Proteins in Urine. Chromatographia 2009, 70, 1473–1478.10.1365/s10337-009-1317-3
- Wu, Y.; Xie, J.; Wang, F.; Chen, Z. Separation of Small Molecular Peptides with Same Amino Acid Composition but Different Sequences by Capillary Electrophoresis. J. Sep. Sci. 2009, 32, 437–440.10.1002/jssc.v32:3
- Hong, N.-S.; Shi, L.H.; Jeong, J.S.; Yang, I.; Kim, S.-K.; Park, S.-R. Rapid and Accurate Determination of Deoxyribonucleoside Monophosphates from DNA Using Micellar Electrokinetic Chromatography with a Cationic Surfactant Additive. Anal. Bioanal. Chem. 2011, 400, 2131–2140.10.1007/s00216-011-4915-5
- Kratschmar, D.; Wallner, S.; Florenski, M.; Schmid, D.; Kuhn, R. Analysis of Oligosaccharides by MEKC with Aminobenzoic Alkyl Esters as Derivatization Agents. Chromatographia 1999, 50, 596–600.10.1007/BF02493666
- Tran, N.T.; Taverna, M.; Deschamps, F.S.; Morin, P.; Ferrier, D. Investigation of Micelles and Anionic Cyclodextrins as Pseudostationary Phases for the Capillary Electrophoresis Separation of Oligosaccharides Derivatized with 2-Amino-Benzamide. Electrophoresis 1998, 19, 2630–2638.10.1002/(ISSN)1522-2683
- Česla, P.; Fischer, J.; Jandera, P. Improvement of the Sensitivity of 2D LC-MEKC Separation of Phenolic Acids and Flavonoids Natural Antioxidants Using the on-Line Preconcentration Step. Electrophoresis 2012, 33, 2646–2473.
- del Mar, C.L.M.; López Vilariño, J.M.; González Rodriguez, M.V.; Barral Losada, L.F. Development, Validation and Application of Micellar Electrokinetic Capillary Chromatography Method for Routine Analysis of Catechins, Quercetin and Thymol in Natural Samples. Microchem. J. 2011, 99, 431–469.
- Liu, Ch-M; Chen, Ch-Y; Lin, Y.-W. Estimation of Tea Catechin Levels Using Micellar Electrokinetic Chromatography: A Quantitative Approach. Food Chem. 2014, 150, 145–150.10.1016/j.foodchem.2013.10.140
- Ye, N.; Li, J.; Wang, Y.; Ma, J. Determination of Catechins in Tea by Micellar Electrokinetic Chromatography with a Graphene Oxide-Coated Capillary. Instrum. Sci. Technol. 2014, 42, 605–617.10.1080/10739149.2014.930876
- Silva, M. Micellar Electrokinetic Chromatography: A Practical Overview of Current Methodological and Instrumental Advances. Electrophoresis 2011, 32, 149–165.10.1002/elps.201000344
- Zhang, Y.; Zhang, R.; Hjertén, S.; Lundahl, P. Liposome Capillary Electrophoresis for Analysis of Interactions between Lipid Bilayers and Solutes. Electrophoresis 1995, 16, 1519–1523.10.1002/(ISSN)1522-2683
- Roberts, M.A.; Locascio-Brown, L.; MacCrehan, W.A.; Durst, R.A. Liposome Behavior in Capillary Electrophoresis. Anal. Chem. 1996, 68, 3434–3440.10.1021/ac9603284
- Bilek, G.; Kremser, L.; Blaas, D.; Kenndler, E. Analysis of Liposomes by Capillary Electrophoresis and Their Use as Carrier in Electrokinetic Chromatography. J. Chromatogr. B 2006, 841, 38–51.10.1016/j.jchromb.2006.03.031
- Wiedmer, S.K.; Hautala, J.; Holopainen, J.M.; Kinnunen, P.K.J.; Riekkola, M.-L. Study on Liposomes by Capillary Electrophoresis. Electrophoresis 2001, 22, 1305–1313.10.1002/(ISSN)1522-2683
- Wiedmer, S.K.; Shimmo, R. Liposomes in Capillary Electromigration Techniques. Electrophoresis 2009, 30, S240–S257.10.1002/elps.v30.10s
- Helle, A.; Mäkitalo, J.; Huhtanen, J.; Holopainen, J.M.; Wiedmer, S.K. Antibiotic Fusidic Acid Has Strong Interactions with Negatively Charged Lipid Membranes: An Electrokinetic Capillary Chromatographic Study. BBA-Biomembranes 2008, 1778, 2640–2647.10.1016/j.bbamem.2008.06.019
- Muhonen, J.; Holopainen, J.M.; Wiedmer, S.K. Interactions between Local Anesthetics and Lipid Dispersion Studied with Liposome Electrokinetic Capillary Chromatography. J. Chromatogr. A 2009, 1216, 3392–3397.10.1016/j.chroma.2009.02.028
- Nijveldt, R.J.; van Nood, E.; van Hoorn, D.E.C.; Boelens, P.G.; van Norren, K.; van Leeuwen, P.A.M. Flavonoids: A Review of Probable Mechanisms of Action and Potential Applications. Am. J. Clin. Nutr. 2001, 74, 418–425.
- Heim, K.E.; Tagliaferro, A.R.; Bobilya, D.J. Flavonoid Antioxidants: Chemistry, Metabolism and Structure-Activity Relationships. J. Nutr. Biochem. 2002, 13, 573–584.
- Özçelik, B.; Kartal, M.; Orhan, I. Cytotoxicity, Antiviral and Antimicrobial Activities of Alkaloids, Flavonoids, and Phenolic Acids. Pharm. Biol. 2011, 49, 396–402.10.3109/13880209.2010.519390
- Bushey, M.M.; Jorgenson, J.W. Separation of Dansylated Methylamine and Dansylated Methyl-D3-Amine by Micellar Electrokinetic Capillary Chromatography with Methanol-Modified Mobile Phase. Anal. Chem. 1989, 61, 491–493.10.1021/ac00180a022
- Wiedmer, S.K.; Lokajová, J.; Riekkola, M.-L. Marker Compounds for the Determination of Retention Factors in EKC. J. Sep. Sci. 2010, 33, 394–409.10.1002/jssc.v33:3
- Laine, J.; Lokajová, J.; Parshintsev, J.; Holopainen, J.M.; Wiedmer, S.K. Interaction of a Commercial Lipid Dispersion and Local Anesthetics in Human Plasma: Implications for Drug Trapping by “Lipid-Sinks”. Anal. Bioanal. Chem. 2010, 396, 2599–2607.10.1007/s00216-009-3435-z
- Lokajová, J.; Laine, J.; Puukilainen, E.; Ritala, M.; Holopainen, J.M.; Wiedmer, S.K. Liposomes for Entrapping Local Anesthetics: A Liposome Electrokinetic Chromatographic Study. Electrophoresis 2010a, 31, 1540–1549.
- Lokajová, J.; Pukkila, J.; Holopainen, J.M.; Wiedmer, S.K. In Vitro Capturing of Various Lipophilic Illicit Drugs by Lipid Dispersions. an Electrokinetic Capillary Chromatography and Fluorescence Polarization Study. Eur. J. Pharm. Sci. 2010b, 41, 515–522.10.1016/j.ejps.2010.08.006
- Chen, N.; Terabe, S.; Nakagawa, T. Effect of Organic Modifier Concentrations on Electrokinetic Migrations in Micellar Electrokinetic Chromatography. Electrophoresis 1995, 16, 1457–1462.10.1002/(ISSN)1522-2683
- Palmer, C.P.; Terabe, S. Micelle Polymers as Pseudostationary Phases in MEKC: Chromatographic Performance and Chemical Selectivity. Anal. Chem. 1997, 69, 1852–1860.10.1021/ac960801d
- Cifuentes, A.; Bernal, J.L.; Diez-Masa, J.C. Determination of Critical Micelle Concentration Values Using Capillary Electrophoresis Instrumentation. Anal. Chem. 1997, 69, 4271–4274.10.1021/ac970696n