
Abstract
Artocarpus hirsutus Lam., one of the species of Artocarpus, is not well studied. The present study is intended to isolate the chemical constituents present in stem wood extract of A. hirsutus, to synthesize their new derivatives and to evaluate their activity against Propionibacterium acnes. Three major flavonoids, cudraflavone A, cycloartocarpin and artocarpin were isolated from the stem wood extract and cudraflavone A was isolated for the first time from this plant. New prenyl ether and acetate derivatives of isolated flavonoids have been synthesized. This is the first to report on the evaluation of in vitro anti-acne potential of crude extracts, isolated flavonoids and their new synthetic derivatives. The crude extracts (MIC values 2, 4 and 15.6 μg/mL) and the isolated constituents (MIC values 2, 0.5 and 0.25 μg/mL) showed significant in vitro anti-acne activity against P. acnes which was comparable with commercially available anti-acne agent, tea tree, oil and a standard antibiotic, Clindamycin (MIC 0.03 μg/mL) indicating their potential as anti-acne agents.
Acne is one of the major skin diseases that are visualized prominently at the age groups 14–20 years. It is recommended to treat acne at their early stages before they form the wounds or further worsen which leads to depression. Despite commercial viability of topical and oral medications in controlling acne, it is significant to discover precise drug molecules possessing better potency, minimal toxicity and capable of specific delivery onto disease site. Medications derived from phytochemicals are becoming reliable due to their safety, tolerability and efficacy. Due to the medicinal value of Artocarpus species, this study is intended to isolate the chemical components present in stem wood extract of A. hirsutus, to synthesize new derivatives and to evaluate their anti-acne potential. Among the isolated compounds, cudraflavone A, cycloartocarpin and artocarpin showed significant in vitro anti-acne activity comparable with commercially available anti-acne agent tea tree oil and standard antibiotic, Clindamycin.
Competing Interests
The authors declare no competing interest.
1. Introduction
Artocarpus hirsutus Lam. plant grows mostly in India, especially in heavy rainfall area of Western Ghat, Kerala and is commonly used as a traditional medicine. It belongs to Moraceae family; studies on other species of this family are well documented (Citation1–16) apart from this plant. Recent few reports (Citation17–19) emphasize the importance of this species and thus it needs to be explored more precisely. Pharmacological studies on bark, root, leaf and fruit extracts have demonstrated their importance as antioxidant agents, as well as their efficacies on diuretic, antibacterial, anti-fungal and antiulcer activity (Citation19, 20). Despite its medicinal values, except our recent report (Citation21) there are no other studies on anti-acne potential of its crude extracts, its isolated chemical constituents or their synthetic derivatives. Acne vulgaris, commonly known as acne, one of the major skin diseases caused by the gram-positive bacteria Propionibacterium acnes (P. acne), appear on human skin as bumps and believed to occur by blockage of skin pores with oils and dead skin cells. Acne vulgaries are visualized prominently at the age groups 14–20 years with extravagant circulation of androgens resulting in enhancement of sebum secretion, a critical inducer in pathogenesis of acne (Citation22). The present study is thus focused to evaluate the in vitro anti-acne potential of crude extracts of leaf, stem bark and stem wood of A. hirsutus. In addition, it is also aimed for the isolation of active components from the extracts, the synthesis of new prenyl ether and acetate derivatives of the actives and evaluation of their in vitro anti-acne potential.
2. Results and discussion
2.1. Chemistry
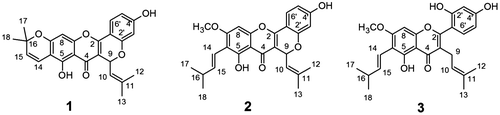
The stem wood powder of A. hirsutus was refluxed with 9 volume of ethanol for 6 h, filtered and dried. It was further soaked with 10 volume of water, stirred at RT for 4 h and filtered to obtain water-soluble and water-insoluble fractions. The MIC of the water-insoluble fraction was found to be better than water-soluble fraction. Thus, water-insoluble fraction was further purified and isolated three flavonoids and characterized as cudraflavone A (1), cycloartocarpin (2) and artocarpin (3) (Figure ), respectively, from their spectroscopic analysis. The purity of compounds 2 and 3 were analysed by HPLC (Supplementary data: Figures S17–S18). The analytical data of compounds 1, 2 and 3 were comparable with the reported values (Citation23–25).
Figure 1. Structures of cudraflavone A (1), cycloartocarpin (2) and artocarpin (3).
2.1.1. Acetates
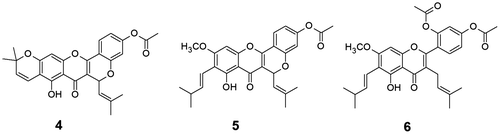
Acetate derivatives are reported to be as active pro-drugs and release drug molecules by hydrolysis, thereby reduce the toxicity as well as enhance the bioactivity of the drugs (Citation26). Cudraflavone A acetate (4), cycloartocarpin acetate (5) and artocarpin diacetate (6) (Figure ) were prepared by acetylation of compound 1, 2 and 3, respectively. New acetate derivatives 4 and 5 were characterized from their respective spectroscopic analysis.
Figure 2. Structures of cudraflavone A acetate (4), cycloartocarpin acetate (5) and artocarpin diacetate (6).
Cudraflavone A acetate (4), a new molecule, was obtained as yellow powder (yield: 47%). 1H NMR spectrum clearly indicated that despite the presence of two phenolic hydroxyl groups in compound 1; only one phenolic OH was derivatized. The disappearance of signal at δ 10.5 ppm (4′-OH) as well as the presence of chelated phenolic hydroxyl signal at δ 12.9 ppm (5′-OH) indicated that phenolic OH at position 4′ was acetylated and 5-OH was remained unchanged. It was also observed that the signals of 6′, 5′ and 3′ protons in acetate derivative, 4 were shifted downfield by ~0.2 to 0.3 ppm compared to the parent compound 1 (Table ). The change in chemical shifts clearly demonstrated that 4′-phenolic OH in compound 4 was acetylated. This result is in good agreement with the reported analytical data of acetate derivative of artocarpin (Citation27). It was further verified by FeCl3 test. The addition of methanolic FeCl3 solution to methanolic solution of compound 4 produced green colour which demonstrated the presence of underivatized-chelated phenolic OH at position 5 in compound 4 (Supplementary data: Figures S19–S21). It was comparable with the reported FeCl3 test of flavonoids possessing chelated phenolic OH at position 5 (Citation27–29). Acetate derivative 4 was assigned with the presence of prominent signals at δ 2.30 (s, 3H, –OCOCH3) in 1H NMR spectrum and an ester carbonyl at δ 169.0 (OCOCH3) in 13C NMR spectrum together with the presence of other characteristic signals. Its molecular mass was confirmed by HRMS. (Supplementary data: Figures S1–S4).
Table 1. 1H NMR (300 MHz) data of 1 in DMSO-d6 and CDCl3 and 4 in CDCl3
Cycloartocarpin acetate (5), a new compound, was obtained as yellow needles with 37% yield. Typical singlet at δ 2.31 (s, 3H, –OCOCH3) together with unaffected phenolic OH signal at δ 13.29 (s, 1H, 5-OH) in 1H NMR spectrum and the presence of single carbonyl signal at δ 168.9 (–OCOCH3) in 13C NMR spectrum demonstrated this acetate derivative 5 as a mono acetate and m/z (HRMS) peak at 477.19120 (M+H)+ is in good agreement with the desired structure. The presence of unreacted phenolic OH at position 5 in compound 5 was further verified by FeCl3 test (Supplementary data: Figures S19–S21).
Similarly, acetylation of artocarpin afforded artocarpin diacetate (6), a known molecule (Citation27), with 59% yield. The presence of diacetates was determined from the characteristic signals at δ 2.33 (s, 3H, –OCOCH3) and 2.12 (s, 3H, –OCOCH3) in 1H NMR spectrum as well as with carbonyl signals at δ 168.8 (OCOCH3) and 168.6 (OCOCH3) in 13C NMR spectrum.
2.1.2. Prenyl ethers
Structural modification of lead natural products is found to be the useful technique to enhance the bioactivity of parent molecules (Citation30). Prenyl ether analogues were reported to possess better bioactivity than their parent compounds (Citation31). Besides acetate derivatives, new prenyl ether analogues 7 and 8 of the major flavonoids; cycloartocarpin (2) and artocarpin (3) were synthesized to examine their efficacy on anti-acne activity. The prenylation reaction was performed by treating compounds 2 and 3 with 3,3-dimethyl allyl bromide in presence of potassium carbonate in acetone. Both prenyl ethers 7 and 8 (Figure ) were characterized from their respective spectroscopic analysis.
Figure 3. Structures of 4′-O-prenyl cycloartocarpin (7) and 2′,4′-di-O-prenyl artocarpin (8).
Prenylation of cycloartocarpin (2) occurred at 4′-OH position as the signal of 5-OH at δ 13.5 (s, 1H) was remained unchanged and afforded 4′-O-prenyl cycloartocarpin (7), a new compound, as yellow needles with 87% yield. The prenyl ether formation was confirmed with the characteristic signals that appeared at δ 5.51–5.42 (m, 2H, H-10), 4.52 (d, 2H, J = 6.6 Hz), 1.93 (s, 3H), 1.70 (s, 3H) and at δ 164.0 (C-4′), 139.2 [C-11 and C–O–CH2–CH=C–(CH3)2], 26.0 [C–4′–O–CH2–CH=C–(CH3)2], 18.4 [C–4′–O–CH2–CH=C–(CH3)2] in 1H NMR and 13C NMR spectra respectively. In HRMS the m/z peak at 503.24287 (M + H)+ is in good agreement with the desired structure. (Supplementary data: Figures S9–S12).
Prenylation of artocarpin (3) provided diprenyl ether 8, a new compound, as yellowish oily liquid with 48% yield. The presence of two prenyl groups was confirmed from the corresponding proton signals that were observed at δ 5.38–5.28 (m, 1H), 5.34–5.30 (m, 1H), 4.56 (d, 2H, J = 6.6 Hz), 4.50 (d, 2H, 6.6 Hz), 1.82 (s, 3H), 1.77 (s, 3H), 1.72 (s, 3H), 1.67 (s, 3H) in 1H NMR spectrum. It was found that signal of 5-OH at δ 13.74 (s, 1H) was remained unaltered, whereas both signals of 4′-OH and 2′-OH were disappeared and thus 4′-OH and 2′-OH were prenylated to afford diprenyl compound 8. The prenylation at 4′-OH and 2′-OH was further confirmed from its HMBC spectrum. In HMBC spectrum (Supplementary data: Figures S22–S24) of compound 8, the signal of chelated phenolic OH at position 5 (δ 13.74) showed a cross peak with δ 159.2 (C-5), however no cross peak was observed with C-5 (δ 159.2) and methylene protons of prenyl groups (either at δ 4.56 or at δ 4.50). Whereas two prenyl methylene protons at δ 4.56 and δ 4.50 showed the cross peaks with C-4′ (δ 161.9) and C-2′ (δ 157.8), respectively. Simultaneously, the cross peaks of C-4′ (δ 161.9) and C-2′ (δ 157.8) with H-6′ (δ 7.23) also represent their correlations. Thus, HMBC spectrum of compound 8 confirms that both prenylation occurred in phenolic OH at positions 2′ and 4′ and not at position 5. The characteristic carbon signals in 13C NMR spectrum also assigned the structure. HRMS showed m/z peak at 573.32172 (M + H)+ which is in good agreement with the desired structure. (Supplementary data: Figures S13–S16).
The presence of underivatized chelated phenolic OH at position 5 in both the molecules 7 and 8 was further verified by FeCl3 test (Supplementary data: Figures S19–S21).
It was noticed that phenolic hydroxyl group at position 5 (5-OH) could not be derivatized (as evidenced by 1H NMR and HMBC spectra as well as positive FeCl3 test with reddish green colouration) either by acetylation or by prenylation even at 60°C, whereas other phenolic hydroxyl groups (2′-OH and 4′-OH) were derivatized readily at room temperature. This could be due to the hydrogen bonding of 5-OH with C-4 carbonyl together with steric interference owing to its flanked position in between C-4 carbonyl and C-6 side chain functionality, which might have played the crucial role to restrain 5-OH from derivatization.
2.2. Anti-acne activity
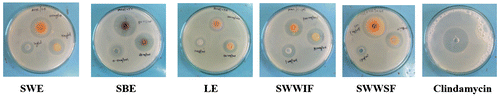
In vitro antibacterial assay was determined by well diffusion method using Reinforced Clostridial Agar M154 (RCA) media under anaerobic conditions. All ethanolic extracts of leaf, stem bark and stem wood of A. hirsutus exhibited significant anti-acne activity and comparable with tea tree oil (Figure ).
Figure 4. Zone of inhibition of crude extracts against P. acnes.
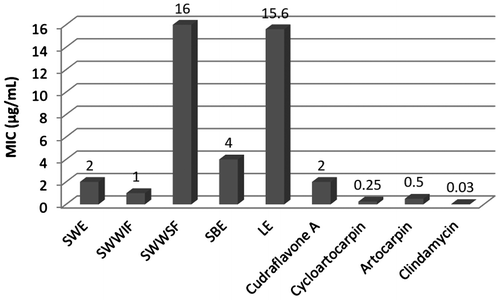
The minimum inhibitory concentration (MIC) of the crude extracts, isolated flavonoids, acetate and prenyl ether derivatives as well as formulated creams were determined by broth micro dilution method. Ethanolic extract of stem wood (MIC = 2 μg/mL) showed 2 and 8-fold more potent against P. acnes than stem bark (MIC = 4 μg/mL) and leaf (MIC = 15.6 μg/mL) extracts, respectively. We aimed on further purification of ethanolic stem wood extract after immobilizing it with water. Two distinct fractions (water-soluble and water-insoluble) were separately collected and examined for their anti-acne activity. In vitro anti-acne potential of water-insoluble fraction (MIC = 1 μg/mL) was found to be 16-fold more potent than water-soluble fraction (MIC = 16 μg/mL). Compounds 1, 2 and 3 isolated from water-insoluble fraction exhibited significantly higher anti-acne activity with MIC values 2, 0.25 and 0.5 μg/mL, respectively (Figure ).
Figure 5. Graphical representation of the MIC values (μg/mL) of crude extracts and isolated flavonoids against P. acnes.
In vitro anti-acne activities of synthetic prenyl ethers and acetates were determined against P. acnes and showed poor activity even up to 100 mg/mL concentration (Table ). Isolated compounds were dissolved in DMSO, whereas the derivatives were dissolved in ethyl acetate owing to their better solubility compared to other solvents. Reduced anti-acne activity of these derivatives may be due to the inability of active compounds to mobilize into the aqueous phase under the experimental condition.
Table 2. MIC value of A. hirsutus extracts against P. acnes
The cream (o/w type) formulations containing artocarpin (2) and cycloartocarpin (3) were prepared in three different concentrations (0.1, 0.5 and 1.0% w/w). The creams containing artocarpin (0.5 and 1.0% w/w) exhibited significantly better P. acnes activity (Table ) compared to cycloartocarpin creams with same composition but the MIC of cycloartocarpin cream could not be determined precisely as it separated out in different phases on dilution with water and thus the erroneous result of cycloartocarpin is not reported herein.
Table 3. MIC values of formulated cream against P. acnes
3. Conclusion
Water insoluble fraction of stem wood extract of the plant A. hirsutus provided three major flavonoids, cudraflavone A (1), cycloartocarpin (2) and artocarpin (3) and to the best of our knowledge, cudraflavone A (1) was isolated for the first time from this plant. Acetates of compounds 1, 2 and prenyl ether derivatives of compounds 2, 3 have been synthesized for the first time. All new molecules were characterized from their spectroscopic analysis. This study is the first to report on the evaluation of in vitro anti-acne potential of crude extracts, isolated constituents 1, 2 and 3, and their new synthetic derivatives. It was revealed that new acetates and prenyl ether derivatives possessed almost negligible activity against P. acnes with respect to their parent molecules. The reduced anti-acne activity of synthesized new derivatives may be due to the inability of active compounds to mobilize into the aqueous phase under the experimental condition. The crude extracts (MIC values 2, 4 and 15.6 μg/mL) and isolated flavonoids 1, 2 and 3 (MIC values 2, 0.25 and 0.5 μg/mL, respectively) exhibited significant in vitro anti-acne activity against P. acnes and the results were comparable with commercially available anti-acne agent, tree tea oil (Table ) and a standard antibiotic, Clindamycin (MIC 0.03 μg/mL) (Figure ). Further studies on compounds 1, 2 and 3 are recommended to determine their commercial application as anti-acne agent for human health benefit.
4. Experimental
4.1. General experimental procedures
1H NMR (300 MHz) and 13C NMR (75 MHz) spectra were recorded on a VARIAN NMR spectrometer. Chemical shifts (δ values) are reported in ppm (parts per million) with respect to TMS as internal standard. DMSO-d6 and CDCl3 were used as solvent. Mass spectrometry (MS) analysis was carried out on Finnigan LCQ Advantage Max (Thermo, LAM 10234). Normal phase TLC was performed on pre-coated silica gel 60 F254 plates (Merck KGaA) and were visualized either by UV (UV-254/366 nm) or exposed to iodine vapours. HPLC analyses were performed using Shimadzu, LC-20AD system. Reversed phase BDS Hypersil C18 (Thermo) (250 × 4.6 cm, 5 μM) column was used for analysis with mobile phase A: 0.05% acetic acid in water; mobile phase B: 100% acetonitrile; flow rate: 1.0 mL/min; detector: PDA at 254 nm; injection volume: 20 μL (sample dissolved in HPLC grade methanol); run time: 30 min.
4.2. Collection of plant material
Fresh samples of leaf, stem bark and stem wood of A. hirsutus were collected from Udupi district, Karnataka, India during the month of January 2014 and were authenticated by Dr. M. D. Rajanna (Professor, Botanical Garden, University of Agricultural Science, GKVK, Bangalore, India). The sample voucher was kept in herbarium (RD/HAR-AH/10, 11 and 12). The plant parts were cut into small pieces and dried under shade. The dried materials were pulverized to coarse powder and stored in air tight containers.
4.3. Extraction and purification
The leaf, stem bark and stem wood powders (3 kg each) of A. hirsutus were refluxed separately with ethanol (w/v ratio 1:10) for 6 h. The ethanolic extracts were filtered and dried completely under vacuum to obtain leaf extract (LE), 120 g; stem bark extract (SBE), 138 g and stem wood extract (SWE), 175 g. All samples were stored separately in air tight containers.
The ethanolic stem wood extract (150 g) of A. hirsutus (SWE) was soaked with 10 volume of water and stirred at room temperature for 4 h and filtered to separate both water-soluble and water-insoluble fractions. Water-insoluble fraction was further purified and isolated compounds 1, 2 and 3. The water-insoluble fraction of stem wood extract (SWWIF) was subjected to silica gel (60–120 mesh) column chromatography. Elution with chloroform:acetone (1:0–1:1) provided five fractions. Similar fractions were combined and concentrated under reduced pressure. Fractions 1 and 2 provided an oily material in very small quantity and could not be studied further. Material resulted from fraction 3 was crystallized with chloroform to yield compound 1 (400 mg) as brown amorphous powder. Crystallization of fraction 4 with chloroform: methanol (1:1) afforded compound 2 (1 g) as yellow powder. Fraction 5 was purified over silica gel and the desired portion was collected and recrystallized with chloroform to obtain compound 3 (2 g) as yellow crystalline powder.
4.4. Preparation of acetate and prenyl ether derivatives
4.4.1. Acetate derivatives of compounds 1, 2 and 3
The acetate derivatives of isolated compounds 1, 2 and 3 were prepared by following reported method (Citation32). In brief, each compound (1 mmol) was dissolved in acetic anhydride (2 mL) and then potassium carbonate (1.2 mmol) was added. The reaction mixture was stirred at room temperature for 20 h and then diluted with ethyl acetate (50 mL). Ethyl acetate layer was washed with saturated NaHCO3 solution (20 mL × 1), water (20 mL × 2) and brine solution (20 mL × 1), respectively. The organic layer was separated, dried over Na2SO4, filtered and concentrated to afford acetate derivatives 4, 5 and 6 (Figure ) respectively.
4.4.2. Prenyl ether derivatives of compounds 2 and 3
The prenyl ether derivatives of compounds 2 and 3 were prepared by following known literature procedure (Citation33). In brief, each compound (1 mmol) was dissolved in 5 mL of acetone and potassium carbonate (1.1 mmol) was added. The reaction mixture was stirred at 0–10°C for 30 min. To this solution, 3,3-dimethyl allyl bromide (1.1 mmol) was added drop wise under stirring at 0–10°C. After completion of addition, the reaction mixture was stirred at room temperature for 4 h and then diluted with ethyl acetate (50 mL). Ethyl acetate layer was washed with cold 1 N HCl solution (20 mL × 1), water (20 mL × 2) and brine solution (20 mL × 1) respectively. The organic layer was separated, dried over Na2SO4, filtered and concentrated to provide 4′-O-prenyl ether derivative 7 and 2′,4′-di-O-prenyl ether derivative 8 respectively (Figure ).
4.5. Analytical data
4.5.1. Cudraflavone A (1)
Brown amorphous powder; yield 0.013%; mp 261–263°C; TLC Rf 0.8 (chloroform:methanol, 9.5:0.5); 1H NMR (DMSO-d6, 300 MHz): δ 13.17 (s, 1H, 5-OH), 10.60–10.50 (bs, 1H, 4′-OH), 7.65 (d, 1H, J = 8.4 Hz, H-2′), 6.59 (d, 1H, J = 10.2 Hz, H-14), 6.57 (dd, 1H, J = 8.4, 2.4 Hz, H-3′), 6.54 (s, 1H, H-8), 6.35 (d, 1H, J = 2.4 Hz, H-5′), 6.13 (d, 1H, J = 9.3 Hz, H-9), 5.79 (d, 1H, J = 9.9 Hz, H-10), 5.40 (d, 1H, J = 9.6 Hz, H-15), 1.89 (s, 3H, H-13), 1.65 (s, 3H, H-12), 1.431 (s, 3H, H-17), 1.425 (s, 3H, H-18); 13C NMR (DMSO-d6: 75 MHz) δ 177.6 (C-4), 163.5 (C-7), 158.4 (C-2), 157.6 (C-2′), 155.7 (C-8a), 155.5 (C-5), 155.48 (C-4′), 138.3 (C-11), 129.0 (C-15), 125.5 (C-6′), 121.0 (C-10), 114.5 (C-14), 110.2 (C-1′), 108.4 (C-3′), 106.4 (C-4a), 104.9 (C-6), 104.8 (C-3), 103.8 (C-5′), 95.1 (C-8), 78.0 (C-16), 68.8 (C-9), 27.8 (C-17), 27.7 (C-18), 25.4 (C-13), 18.4 (C-12); APCI–MS m/z 418.96 (M + H+) and 417.41 (M − H−), (C25H22O6 requires 418.4385), Lit. (Citation23).
4.5.2. Cycloartocarpin (2)
Pale yellow powder; yield 0.033%; mp 253–255°C; TLC Rf 0.66 (9.5:0.5; chloroform:methanol); HPLC purity 99% (Figure S17). 1H NMR (DMSO-d6, 300 MHz): δ 13.54 (s, 1H, 5-OH), 10.54 (s, 1H, 4′-OH), 7.67 (d, 1H, J = 8.4 Hz, H-6′), 6.79 (s, 1H, H-8), 6.59 (dd, 1H, J = 16.20, 6.6 Hz, H-15), 6.56 (dd, 1H, J = 8.4, 2.1 Hz, H-5′), 6.44 (d, 1H, J = 16.20 Hz, H-14), 6.34 (d, 1H, J = 2.1 Hz, H-3′), 6.12 (d, 1H, J = 9.3 Hz, H-9), 5.34 (d, 1H, J = 9.3 Hz, H-10), 3.90 (s, 3H, 7-OCH3), 2.48 (m, 1H, H-16), 1.87 (s, 3H), 1.64 (s, 3H), 1.03 (d, 6H, J = 6.6 Hz, H-17, H-18); 13C NMR (DMSO-d6, 75 MHz): δ 178.5 (C-4), 164.2 (C-4′), 162.9 (C-7), 158.7 (C-5), 158.3 (C-2′), 156.1 (C-2), 155.5 (C-8a), 142.2 (C-15), 139.0 (C-11), 126.2 (C-6′), 121.7 (C-10), 116.4 (C-14), 110.9 (C-5′), 109.4 (C-1′), 109.2 (C-6), 107.1 (C-3), 105.2 (C-4a), 104.5 (C-3′), 91.4 (C-8), 69.6 (C-9), 57.1 (–OCH3), 33.2 (C-16), 26.1 (C-13), 23.3 (C-17; C-18), 19.1 (C-12); APCI–MS m/z 434.91 (M + H+) and 433.10 (M − H−), (C26H26O6 requires 434.4810), Lit. (Citation24).
4.5.3. Artocarpin (3)
Yellow crystalline powder, yield 0.066%; mp 160–162°C; TLC Rf 0.35 (chloroform:methanol, 9.5:0.5); HPLC purity 96% (Figure S18). 1H NMR (DMSO-d6, 300 MHz): δ 13.87 (s, 1H, 5-OH), 9.90 (s, 1H, 4′-OH), 9.79 (s, 1H, 2′-OH), 7.09 (d, 1H, J = 8.1 Hz, H-6′), 6.64 (s, 1H, H-8), 6.63 (dd, 1H, J = 15.3, 8.4 Hz, H-15), 6.48 (d, 1H, J = 15.6 Hz, H-14), 6.42 (d, 1H, J = 2.4 Hz, H-3′), 6.33 (dd, 1H, J = 8.1, 2.4 Hz, H-5′), 5.02 (t, 1H, J = 6.0 Hz, H-10), 3.86 (s, 3H, 7-OCH3), 2.98 (d, 2H, J = 6.0 Hz, H-9), 2.45–2.33 (m, 1H, H-16), 1.53 (s, 3H, H-12), 1.35 (s, 3H, H-13), 1.04 (d, 6H, J = 6.6 Hz, H-17 and H-18); 13C NMR (DMSO-d6, 75 MHz): δ 182.5 (C-4), 163.2 (C-7), 162.7 (C-8a), 161.1 (C-4′), 158.7 (C-5), 157.1 (C-2′), 156.6 (C-2), 141.9 (C-15), 132.0 (C-11), 131.8 (C-6′), 122.1 (C-3), 120.9 (C-10), 116.5 (C-14), 111.5 (C-1′), 108.8 (C-6), 107.4 (C-5′), 104.8 (C-4a), 103.3 (C-3′), 90.8 (C-8), 57.1 (7-OCH3), 33.2 (C-16), 26.1 (C-12), 24.3 (C-9), 23.3 (C-17; C-18), 18.0 (C-13); APCI–MS m/z 437.13 (M + H+) and 435.10 (M-H-), (C26H28O6, requires 436.4969), Lit. (Citation25).
4.5.4. Cudraflavone A acetate (4)
Yellow power; yield 47%; mp 203–206°C; TLC Rf 0.57 (hexane:ethyl acetate, 4:1).
13C NMR (CDCl3, 75 MHz): δ 178.9 (C-4), 169.0 (OCOCH3), 159.5, 157.4, 156.7, 156.6, 155.0, 154.4, 139.9, 128.4, 124.8, 121.1, 115.7, 115.3, 113.6, 111.6, 111.5, 106.0, 105.9, 95.4 (C-8), 78.2 (C-16), 70.13 (C-9), 28.5, 28.45, 26.1, 21.4 (OCOCH3), 18.9; APCI MS m/z 460.97 (M + H+); HRMS Calcd. for C27H25O7 (M + H)+: 461.15948; found: 461.15977. (Figures S1–S4).
4.5.5. Cycloartocarpin acetate (5)
Yellow needles; yield 37%; mp 159–161°C; TLC Rf 0.37 (4:1, hexane:ethyl acetate); 1H NMR (CDCl3, 300 MHz): δ 13.29 (s, 1H, 5-OH), 7.75 (d, 1H, J = 8.7 Hz, H-6′), 6.81 (dd, 1H, J = 8.7, 2.1 Hz, H-5′), 6.71 (d, 1H, J = 2.4 Hz, H-3′), 6.69 (dd, 1H, J = 16.2, 6.6 Hz, H-15), 6.55 (d, 1H, J = 16.5 Hz, H-14), 6.45 (s, 1H, H-8), 6.27 (d, 1H, J = 9.6 Hz, H-9), 5.43 (d, 1H, J = 9.6 Hz, H-10), 3.93 (s, 3H, 7-OCH3), 2.53–2.42 (m, 1H, H-16), 2.31 (s, 3H, –OCOCH3), 1.97 (s, 3H, H-12), 1.70 (s, 3H, H-13), 1.11 (d, 6H, J = 6.9 Hz, H-17 and H-18); 13C NMR (CDCl3,, 75 MHz): δ 178.9 (C-4), 168.9 (–OCOCH3), 162.9, 159.2, 157.3, 155.4, 155.0, 154.2, 143.0, 139.8, 124.6, 121.1, 115.7, 113.5, 113.5, 111.8, 111.5, 110.3, 106.0, 89.9 (C-8), 70.2 (C-9), 56.2 (OCH3), 33.3, 26.1, 22.88, 22.86, 21.4 (OCOCH3), 18.9; APCI-MS m/z 475.12 (M − H−); 477.03 (M + H)+; HRMS Calcd. for C28H29O7 (M + H)+: 477.19078; found: 477.19120. (Figures S5–S8).
4.5.6. Artocarpin Diacetate (6)
Yellow powder; yield 59%; mp 153–155°C; TLC Rf 0.31 (hexane:ethyl acetate, 4:1); 1H NMR (CDCl3, 300 MHz): δ 13.50 (s, 1H, 5-OH), 7.48 (d, 1H, J = 8.7 Hz, H-6′), 7.16–7.11 (m, 2H, H-3′, H-5′), 6.73 (dd, 1H, J = 16.2, 6.9 Hz, H-15), 6.58 (d, 1H, J = 16.2 Hz, H-14), 6.31 (s, 1H, H-8), 5.13–5.07 (m, 1H, H-10), 3.89 (s, 3H, 7-OCH3), 3.07 (d, 2H, J = 6.3 Hz, H-9), 2.55–2.45 (m, 1H, H-16), 2.33 (s, 3H, –OCOCH3), 2.12 (s, 3H, –OCOCH3), 1.63 (s, 3H, H-13), 1.43 (s, 3H, H-12), 1.12 (d, 6H, J = 6.6 Hz, H-17, H-18); 13C NMR (CDCl3, 75 MHz): δ 182.3 (C-4), 168.8 (–OCOCH3), 168.6 (–OCOCH3), 163.3 (C-7), 159.2 (C-5), 158.2 (C-2), 156.3 (C-8a), 152.7 (C-2′), 149.1 (C-4′), 142.9 (C-15), 133.1 (C-6′), 131.2 (C-11), 123.6 (C-10), 121.9 (C-3), 121.1 (C-1′), 119.5 (C-5′), 117.3 (C-3′), 115.8 (C-14), 110.0 (C-6), 105.3 (C-4a), 89.3 (C-8), 56.2 (–OCH3), 33.4 (C-16), 25.9 (C-9), 24.3 (C-12), 23.0 (–OCOCH3), 23.0 (–OCOCH3), 21.4 (C-17), 21.0 (C-18), 17.9 (C-13); APCI-MS m/z 521.16 (M + H+), (C30H32O8 requires 520.5703), Lit. (Citation27).
4.5.7. 4′O-Prenyl cycloartocarpin (7)
Yellow needles; yield 87%; mp 122–126°C; TLC Rf 0.76 (hexane:ethyl acetate, 4:1); 1H NMR (CDCl3, 300 MHz): δ 13.5 (s, 1H, 5-OH), 7.7 (d, 1H, J = 8.7 Hz, H-6′), 6.71 (dd, 1H, J = 16.20, 6.9 Hz, H-15), 6.60 (dd, 1H, J = 8.9, 2.4 Hz, H-5′), 6.56 (d, 1H, J = 16.20 Hz, H-14), 6.47 (d, 1H, J = 2.4 Hz, H-3′), 6.43 (s, 1H, H-8), 6.26 (d, 1H, J = 9.3 Hz, H-9), 5.51–5.42 (m, 2H, H-10 and 4′–O–CH2–CH=), 4.52 (d, 2H, J = 6.6 Hz, 4′–O–CH2–CH=), 3.92 (s, 3H, 7-OCH3), 2.53–2.42 (m, 1H, H-16), 1.93 [s, 3H, 4′–O–CH2–CH=C–(CH3)2], 1.81 (s, 3H), 1.75 (s, 3H), 1.70 [s, 3H, 4′–O–CH2–CH=C–(CH3)2], 1.11 (d, 6H, J = 6.9 Hz, H-17 and H-18); 13C NMR (CDCl3, 75 MHz): δ 178.7 (C-4), 164.0, 162.6, 159.3, 158.2, 155.4, 155.4, 142.8, 139.2, 125.0, 121.3, 119.0, 115.9, 110.0, 109.7, 108.6, 105.9, 103.0, 89.9 (C-8), 70.1 (C-9), 65.3 [–O–CH2–CH=C–(CH3)2], 56.1 (–OCH3), 33.4, 26.2, 26.0, 22.9, 22.9, 18.9, 18.4; APCI MS m/z 501.18 (M − H−); HRMS Calcd. for C36H35O6 (M + H)+: 503.24282, found: 503.24287. (Figures S9–S12).
4.5.8. 2′,4′-Di-O-prenyl artocarpin (8)
Yellowish oily liquid; yield 48%; TLC Rf 0.80 (hexane:ethyl acetate, 4:1); 1H NMR (CDCl3, 300 MHz): δ 13.74 (s, 1H, 5-OH), 7.23 (d, 1H, J = 9.0 Hz, H-6′), 6.72 (dd, 1H, J = 16.2, 6.6 Hz, H-15), 6.74–6.56 (m, 3H, H-3′, H-5′, H-14), 6.33 (s, 1H, H-8), 5.38–5.28 [m, 1H, –O–CH2–CH=C–(CH3)2], 5.34–5.30 [m, 1H, –O–CH2–CH=C–(CH3)2], 5.11–5.08 (m, 1H, H-10), 4.56 [d, 2H, J = 6.6 Hz, –O–CH2–CH=C–(CH3)2], 4.50 [d, 2H, 6.6 Hz, –O–CH2–CH=C–(CH3)2], 3.05 (d, 2H, J = 6.9 Hz, H-9), 2.51–2.45 (m, 1H, H-16), 1.82, 1.77, 1.72, 1.67 [4s, 12H, –O–CH2–CH=C–(CH3)2], 1.60 (s, 3H), 1.41 (s, 3H), 1.11 (d, 6H, J = 6.9 Hz, H-17 and H-18); 13C NMR (CDCl3, 75 MHz): δ 182.7 (C-4), 162.8, 161.9, 160.8, 159.2, 157.8, 156.6, 142.4, 139, 137.9, 132.07, 131.5, 121.9, 121.6, 119.6, 119.4, 116.1, 115.3, 109.4, 105.6, 105.5, 100.8, 89.5 (C-8), 65.8 and 65.2 [–O–CH2–CH=C–(CH3)2], 56.1 (OCH3), 33.4, 31.2, 26.l, 25.94, 25.87, 24.5, 23.0 18.5, 17.8; APCI-MS m/z 573.17 (M + H+); HRMS Calcd. for C36H45O6 (M + H)+: 573.32107, found: 573.32172. (Figures S13–S16).
4.6. Formulation of compounds 2 and 3
Creams (o/w type emulsion) with varying concentrations of compounds 2 and 3 (0.1, 0.5 and 1.0% w/w) were prepared as follows: The cream base was prepared by adding phase A containing arlacel 165, cetyl alcohol, Imex IN3, cresmer EW, Capric caprylic triglycerides, Kokum butter and Tinogard TT into the phase B containing tetrasodium EDTA and biopol U10 in water, at 75–80°C on water bath slowly stirring and homogenized at 10,000 rpm. Once the cream base was prepared, the temperature was reduced to 40–45°C. AMP 95 solution in Demineralized (DM) Water, Compounds 2/3 solution in propylene glycol, Xiameter PMX3021, Neolone PE, Vitamin E acetate and perfume were added slowly one by one and homogenized. Finally, the creams were transferred in tubes, sealed and labelled.
4.7. Anti-acne potential
In vitro anti-acne activity of ethanolic leaf, stem bark and stem wood extracts of A. hirsutus, isolated components from stem wood extract, their synthetic derivatives and formulated creams were evaluated against acne-causing bacterium, P. acnes. The antibacterial activity was evaluated by agar well diffusion method. The minimum inhibitory concentrations (MIC) of all compounds were determined by broth micro dilution method.
4.7.1. Microorganisms
Acne causing bacterium P. acnes (ATCC 11827) was procured from American Type Culture Collection, Rockville, USA.
4.7.2. Media
Reinforced Clostridial Agar (Hi Media; M154) and Reinforced Clostridial broth (Hi Media; M443) procured from Hi-media were used as media in the experiments.
4.7.3. Determination of antibacterial activity
The crude ethanolic extracts and isolated compounds were dissolved in DMSO with a concentration range of 0.5–100 mg/mL. Each derivative was dissolved in ethyl acetate to achieve concentration in the range of 100–6.25 mg/mL. The formulated creams were dissolved in water to prepare a concentration up to 4,000 –7.86 μg/mL. P. acnes was cultured on Reinforced Clostridial Agar (RCA) M154 and incubated at 37°C for 48 h in an anaerobic chamber providing gas mixture containing 80% nitrogen, 10% carbon dioxide and 10% hydrogen. The bacterial culture was suspended in sterile normal saline and adjusted to 1.0 × 108 cfu/mL (Citation34). Sterile RCA was seeded with the standardized culture of P. acnes and poured into plates. The agar medium was allowed to solidify. Wells of 7 mm diameter equidistant from each other were punched into the agar surface using a sterile borer. Aliquot of each sample, diluent control (DMSO), ethyl acetate and Clindamycin as standard antibiotic were loaded in the wells. The plates were kept at 4–8°C for 3 h and then incubated in the anaerobic chamber for 48 h. The diameter of zone of inhibition around the wells was measured and recorded (Figure ).
4.7.4. Determination of minimum inhibitory concentration
The minimum inhibitory concentrations of the test samples described above against P. acnes were determined by broth micro dilution method after solubilising them in respective solvents. Two-fold dilutions of the extracts, isolated compounds were prepared in RCB acquiring concentrations in the range of 0.1–2,000 μg/mL. Concentrations of the formulated creams were prepared in the range of 31.25–2,000 μg/mL. The diluted samples were loaded in 96-well micro titre plate. The samples were inoculated with the test culture of P. acnes and maintained the final concentration of the bacteria in each well at 1 × 105 cfu, The P. acnes culture was adjusted to 0.5 McFarland corresponding to a turbidity of 1 to 2 × 108 cfu/mL. This stock solution was diluted with RCB to 1 × 106 cfu/mL. From this 100 μL was added to the wells to get the final concentration of bacteria 1 × 105 cfu/well) (Citation35). The plates were incubated under anaerobic conditions at 37°C for 48 h and monitored for inhibition of bacterial growth.
Supplemental data
1H, 13C NMR, low and high resolution mass spectra of new compounds 4, 5, 7 and 8. HMBC spectrum of compound 8. HPLC chromatograms of compounds 2 and 3. Photographs of FeCl3 test results of compounds 4, 5, 7 and 8.
Supplementary material
Supplemental data for this article can be accessed at https://doi.org/10.1080/23312009.2017.1416557.
Funding
The author(s) thank Sami Labs Limited (SLL), Bangalore, India, for the financial support.
Supplementary_Material.pdf
Download PDF (2.6 MB)Acknowledgements
The author(s) thank Dr N. Kalyanam, Dr S. Natarajan, Dr Samuel Manoharan T., Dr Rajendran R., Mr Anand Desai and Analytical team of SLL for their valuable support.
Additional information
Notes on contributors
A. Nagarajan
A. Nagarajan is the Vice president R&D of Sami Labs and associated with the company for the last 22 years. He obtained his PhD in Organic Chemistry from the University of Madras. He has three years post-doctoral research experience at the University of Oklahoma, USA in the area of organometallic and organic chemistry. His research interests include synthetic methodology, synthesis of chiral molecules and bioactive natural products and to study their significance on biological system. He has more than 30 years of research experience in the field of Organic Chemistry and has published more than 15 articles in reputed journals.
Related Research Data
References
- Abdullah, S.A.; Jamil, S.; Basar, N.; Abdul Lathiff, S.M.; Mohd Arriffin, N. Nat. Prod. Res. 2017, 31, 1113.10.1080/14786419.2016.1222387
- Daud, M.N.H.; Fatanah, D.N.; Abdullah, N.; Ahmad, R. Food Chem. 2017, 232, 621.10.1016/j.foodchem.2017.04.018
- Sun, G.; Zheng, Z.; Lee, M.H.; Xu, Y.; Kang, S.; Dong, Z.; Wang, M.; Gu, Z.; Li, H.; Chen, W. J. Agric. Food Chem. 2017, 65, 3474.10.1021/acs.jafc.7b00278
- Yuan, W.J.; Yuan, J.B.; Peng, J.B.; Ding, Y.Q.; Zhu, J.X.; Ren, G. Fitoterapia 2017, 117, 133.10.1016/j.fitote.2017.01.016
- Arai, M.A.; Uchida, K.; Sadhu, S.K.; Ahmed, F.; Koyano, T.; Kowithayakorn, T.; Ishibashi, M. Bioorg. Med. Chem. 2015, 23, 4150.10.1016/j.bmc.2015.06.058
- Jagtap, U.B.; Bapat, V.A. J. Ethnopharmacol. 2010, 129, 142.10.1016/j.jep.2010.03.031
- Barik, B.R.; Bhaumik, T.; Dey, A.K.; Kundu, A.B. Phytochemistry 1994, 35, 1001.10.1016/S0031-9422(00)90655-1
- Hashim, N.; Rahmani, M.; Sukari, M.A.; Ali, A.M.; Alitheen, N.B.; Go, R.; Ismail, H.B. J. Asian Nat. Prod. Res. 2010, 12, 106.10.1080/10286020903450411
- Hakim, E.H.; Fahriyati, A.; Kau, M.S.; Achmad, S.A.; Makmur, L.; Ghisalberti, E.L.; Nomura, T. J. Nat. Prod. 1999, 62, 613.10.1021/np980279 l
- Makmur, L.; Achmad, S.A.; Aimi, N.; Hakim, E.H.; Kitajima, .M.; Takayama, H. J. Nat. Prod. 2000, 63, 243.10.1021/np990220u
- Wongkham, S.; Wongkham, C.; Boonsiri, P.; Simasathiansophon, S.; Trisonthi, C.; Atisook, K. Phytochemistry 1995, 40, 1331.10.1016/0031-9422(95)00535-F
- Zhang, P.Z.; Gu, J.; Zhang, G.L. J. Asian Nat. Prod. Res. 2015, 17, 217.10.1080/10286020.2015.1006202
- Daus, M.; Chaithada, P.; Phongpaichit, S.; Watanapokasin, R.; Carroll, A.R.; Mahabusarakam, W. Phytochem. Lett. 2017, 19, 226.10.1016/j.phytol.2017.01.007
- Radwan, M.M.; Rodriguez-Guzman, R.; Manly, S.P.; Jacob, M.; Ross, S.A. Phytochem. Lett. 2009, 2, 141.10.1016/j.phytol.2009.04.002
- Lan, W.C.; Tzeng, C.W.; Lin, C.C.; Yena, F.L.; Ko, H.H. Phytochemistry 2013, 67, 824.
- Lin, K.W.; Wang, B.W.; Wu, C.M.; Yen, M.H.; Wei, B.L.; Hung, C.F.; Lin, C.N. Phytochem. Lett. 2015, 14, 190.10.1016/j.phytol.2015.10.013
- Nayak, M.; Nagarajan, A.; Majeed, M.; Lakshmi, A.M. IJSTR 2017, 6, 196.
- Nayak, M.; Nagarajan, A.; Majeed, M. Pharmacog. J. 2017, 9, 887.10.5530/pj
- Shanmugapriya, K.; Akanya, S.; Anderson, C.B.; Berril, N.S.; Prakash, P.R.; Suganya, V. Asian J. Pharm. Clin. Res. 2017, 10, 298.
- Dibinlal, D.; Seethadevi, B. J. Pharma Search 2013, 8, 3.
- Nayak, M.; Nagarajan, A.; Majeed, M.; Kalyanam, N.; Choudhury; A.K. Nat. Prod. Res. Published online August 18, 2017. doi:10.1080/14786419.2017.1365068.
- Lone, A.H.; Habib, S.; Ahmad, T.; Anwar, M. J. Ayurveda Integr. Med. 2012, 3, 180.10.4103/0975-9476.104430
- Wei, B.L.; Weng, J.R.; Chiu, P.H.; Hung, C.F.; Wang, J.P.; Lin, C.N. J. Agric. Food Chem. 2005, 53, 3867.10.1021/jf047873n
- Likhitwitayawuid, K.; Sritularak, B.; De-eknamul, W. Planta Med. 2000, 66, 275.10.1055/s-2000-8656
- Hui, H.T.; Liu, M.F.; Lin, L.D.; Wei, X.Y. J. Trop. Subtrop. Bot. 2012, 20, 529.
- Basmadjian, C.; Zhao, Q.; Bentouhami, E.; Djehal, A.; Nebigil, C.G.; Johnson, R.A.; Serova, M.; de Gramont, A.; Faivre, S.; Raymond, E.; Désaubry, L.G. Front Chem. 2014, 2, 20.
- Cunha, M.P.S.C.; Pinto, A.C.; Braz-Filho, R. J. Braz. Chem. Sco. 1994, 5, 101.10.5935/0103-5053.19940017
- Briggs, L.H.; Locker, R.H. J. Chem. Soc. 1951, 3136–3142.
- Brahmachari, G.; Sohel, S.M.A.; Gorai, D.; Mondal, S.; Misrti, B. J. Chin. Chem. Soc. 2003, 50, 325–328.10.1002/jccs.v50.2
- Yao, H.; Liu, J.; Xu, S.; Zhu, Z.; Xu, J. Expert Opin. Drug Discov. 2017, 12, 121.10.1080/17460441.2016.1272757
- Hairani, R.; Mongkol, R.; Chavasiri, W. Bioorg. Med. Chem. Lett. 2016, 26, 5300.10.1016/j.bmcl.2016.09.044
- Fiorito, S.; Epifano, F.; Taddeo, V.A.; Genovese, S.; Azelmat, J.; Grenier, D. Planta Med. 2016, 81 (S01), S1–S381.
- Fulgentius, N.L.; Koonj, S.; Edwin, H. Catalysts 2013, 3, 954.
- Clinical and laboratory standards Institute (CLSI). 12th ed., M02-A11, 2015, 32(1).
- Clinical and laboratory standards Institute (CLSI), 8th ed., M11-A8, 2012, 32(5).