
Abstract
pH and pKa’s are important parameters in enzyme technology, because they determine the protonated states of titratable groups and thus influence the structure, dynamics, and functions of proteins. Specifically, experimental studies of Thermomyces lanuginosus SSBP revealed the presence of putative family 18 chitinases are triggered by low pHs. In this work, we performed several pH-based molecular dynamics simulations of chitinase II in a water solvent. This work was aimed at establishing the optimum activity and stability profiles of chitinase II. We observed a strong conformational pH dependence of chitinase II and the enzyme retained their characteristic TIM barrel topology at low pH.
Public Interest Statement
In this work, we performed several pH-based Molecular Dynamics (MD) simulations of chitinase II in a water solvent. This work was aimed at establishing the optimum activity and stability profiles of chitinase II. The combination of the Generalized Born and Iterative Mobile Clustering approaches was utilized for the protein ionization and pKa calculations. Titration curves for each one of the titratable amino acid residues present in chitinase II were calculated. The MD simulations at various pHs in an explicit water environment were performed to establish the optimum activity and stability profiles of chitinase II.
Competing interests
The authors declare no competing interest.
1. Introduction
Chitinases are a group of enzymes that hydrolyze the naturally occurring polysaccharide chitins, a linear polymer of β (1,4) N-acetyl β-D-glucosamine (Henrissat, Citation1999). Chitinases are ubiquitous and present in all forms of life including bacteria, fungi, plants, animals, and also in viruses (Khan, Bisetty, Singh, Permaul, & Hassan, Citation2015; Tran, Barnich, & Mizoguchi, Citation2011; Vaaje-Kolstad, Houston, Riemen, Eijsink, & van Aalten, Citation2005). They form a class of extremely conserved enzymes and exhibit varied functions in these organisms. Chitinases possess an extraordinary ability to hydrolyze the highly insoluble chitin polymer directly to a lower molecular weight chitooligomer, widely used in agricultural, biotechnological, industrial, and medical fields (Di Giambattista, Federici, Petruccioli, & Fenice, Citation2001).
The thermostable nature of chitinase II isolated from thermophilic organism Thermomyces lanuginosus has been the primary focus of research in the past few years mainly due to their important biophysiological functions and applications, ranging from biocontrol of fungal phytopathogens (Hamid et al., Citation2013; Singh, Madlala, & Prior, Citation2003) to chemical fungicides due to their inherent stability at high temperatures and wide range of pH (Herrera-Estrella & Chet, Citation1999; Maheshwari, Bharadwaj, & Bhat, Citation2000). Here, we studied two forms of chitinase II for computational studies: one with the signal peptide and the other one without signal peptide.
The knowledge of the protein three-dimensional (3D) structures is crucially important for rational drug design (OuYang et al., Citation2013). Although X-ray crystallography is a powerful tool in determining protein 3D structures, it is time-consuming and expensive, and not all proteins can be successfully crystallized (Berardi, Shih, Harrison, & Chou, Citation2011). To acquire the structural information, 3D protein structures were developed by means of homology technique were used further for pH-based study (Khan, Wei, Gu, Hassan, & Tabrez, Citation2016).
The pH determines the charges of the ionizable residues, exhibiting a direct influence on the intra-molecular electrostatic interactions thereby affecting the molecular structure (Flanagan, Ackers, Matthew, Hanania, & Gurd, Citation1981). The pH-dependent protein stability is a critical factor for protein folding which is key to various molecular mechanisms. In protein folding, the titratable amino acids in the proteins are modified, depending upon the nature of the buried amino acids with no exposure to the solvent. Specifically, the pKa values of the amino acid side chains play a pivotal role in defining the pH-dependent characteristics of a protein (Song, Mao, & Gunner, Citation2009).
Literature studies revealed that pH has a dramatic impact on biomolecules (Matthew et al., Citation1985; Yang & Honig, Citation1993); therefore, pH-dependent Molecular Dynamics (MD) simulations would provide a deeper insight into the structural and functional aspects of the enzyme (Gramany et al., Citation2015). Many remarkable biological functions in proteins and their profound dynamic mechanisms can be revealed by studying their internal motions (Anwer et al., Citation2016; Khan, Aamir, Wei, Ahmad, & Hassan, Citation2016; Khan et al., Citation2016). In order to understand the detailed action mechanism of receptor–ligand binding, the dynamical information obtained by simulating their internal motions or dynamic process is very useful (Anwer et al., Citation2015; Khan, Govender, Permaul, Singh, & Bisetty, Citation2015; Stephens et al., Citation2014).
Accordingly, the MD simulations were performed with a fixed protonation state of the enzyme. The pKa values of the titratable groups were obtained by modulating the protonation states of the various titratable functional groups present in the system (Nielsen & McCammon, Citation2003). The generated changes in charge distribution showed a profound impact on the structure and function of the enzymes. The pH-based MD simulations were performed in an explicit water solvent. The prime objective of this study was to evaluate the effect of the physiological pH on chitinase II enzyme obtained from T. lanuginosus SSBP. This study was aimed at establishing a relationship between physiological pH with the structural and functional aspects of chitinase II. The results demonstrate the influence of pH on the protein structure and dynamics.
2. Materials and methods
2.1. Molecular modeling
The full-length gene for the novel chitinase II derived from T. lanuginosus SSBP was sequenced and submitted to the GeneBank database under accession numbers KJ740647. Since 3D structures are important for the identification of the folding, they were utilized as input structures for the pH calculations and MD simulations. For this purpose, the various modules of Discovery Studio (DS) 4.0 (Eswar, Eramian, Webb, Shen, & Sali, Citation2008) were utilized to predict the 3D model of chitinase II. The model was constructed by reducing the infringements of all the restraints.
2.2. Protein ionization and pKa calculations
The pH-dependent activity and protein stability displayed by enzymes are the properties determined by the pKa values of amino acid side chains. The combination of the Generalized Born and Iterative Mobile Clustering approaches provided new computational protocols for pKa calculations. The pKa and titration curves for each titratable amino acid residues present in chitinase II were calculated using DS 4.0 (Spassov & Yan, Citation2008). On the other hand, Chemistry at HARvard Molecular Mechanics (CHARMm) is a highly versatile and widely used molecular simulation program that primarily focuses on biological molecules such as proteins and peptides. The structures for chitinase II were prepared using the CHARMm Polar hydrogen force field. CHARMm is a program for macromolecular dynamics and mechanics. It performs standard MD using state-of-the-art algorithms for time stepping, long-range force calculation, and periodic images. CHARMm can also be used for energy minimization, normal modes, and crystal optimizations. The total charge of the protein as a function of pH and electrostatic contribution to the free energy as a function of pH were calculated in this work. The isoelectric points (pI) of the chitinase II were predicted at a particular pH and the information can be utilized for precipitation and purification of protein.
2.3. A pH-based MD simulations
The changes in the protonation states were performed using the pdb2gmx module of GROMACS packages (Van Der Spoel et al., Citation2005) by utilizing the information obtained from DS. The generated protonation states for chitinase II were used as inputs for MD simulations at a pH ranging from 2 to 6. MD simulations were performed at a molecular mechanics level using the all atom optimized potential for liquid simulations (OPLS) function. The OPLS-AA (all atom) force field consists of a series of modifications of torsional parameters incorporated into the ab initio results used in the prediction of side-chain rotamer preferences, which are critical for accurate protein homology modeling protocols (Lopes et al., Citation2013). The OPLS-AA force field incorporated into the GROMACS simulation package includes all atoms explicitly modeled and designed to reproduce experimental properties. The OPLS placed a strong emphasis on deriving non-bonded interactions. The full-length chitinase II were soaked in a cubic box of water molecules with a dimension of 10 Å using the “editconf” module for creating boundary conditions and the “genbox” module for solvation. The “spc216” template was used to solvate the protein. The charges on the protein were neutralized using the verlet cut-off scheme by the addition of Na+ and Cl− ions to maintain neutrality. The system was then minimized in order to combat inappropriate geometry and structure distortions using 1,500 steps of steepest descent. This process was executed until a maximum force (<1,000 kJ/mol/nm) was reached.
The temperatures of all the systems were subsequently raised from 0 to 300 K during their equilibration period (100 ps) at a constant volume under periodic boundary conditions. The equilibration was achieved in two phases using the NVT and NPT ensembles (constant number of particles, volume, and temperature at 100 ps). After the equilibration phase, the Particle-Mesh Ewald summation method (Horng, Tracz, Lumb, & Raleigh, Citation2005) was applied, and the production phase consisting of 20 ns were performed. The resulting trajectories were analyzed using g_gyrate, g_rms, g_rmsf, and g_energy utilities of GROMACS. The outputs were analyzed using X-MGRACE (GRaphing Advanced Computation and Exploration of data) plotting tools. All the graphic presentations were prepared using DS 4.0 and Visual MD (Humphrey, Dalke, & Schulten, Citation1996).
2.4. MD analysis
Root mean square deviation (RMSD) is one of the most important fundamental properties for analysis whether the protein is stable and close to the experimental structure (Kuzmanic & Zagrovic, Citation2010). The RMSD is a measure of the deviation of the conformational stability of the proteins from backbone structure to the initial starting structure (Kuzmanic & Zagrovic, Citation2010).
Vibrations around the equilibrium are not random, but depend on local structure flexibility. To calculate the average fluctuation of all residues during the simulation, the root mean square fluctuation (RMSF) of the Cα atoms of protein from the primary structure was plotted as a function of residue number using g_rmsf module of GROMACS (Kuzmanic & Zagrovic, Citation2010).
The Radius of gyration (Rg) analysis provided insight into the compactness of the protein during the simulations. Rg is defined as the root mean square distance of the collection of atoms from their common centre of gravity. If a protein is folded in stable configuration, it will likely maintain a relatively steady value of Rg.
3. Results and discussion
3.1. 3D structure
The 3D structures of chitinases II showed well-defined (β/α)8 TIM Barrel fold . All TIM barrel conformations known to date are enzymes which possess catalytic functions. The predicted models showed overhanging sequences due to the presence of signal peptide. The chitinase II are predicted to have two β-sheets which comprises eight parallel strands, two anti-parallel strands, one β-hairpin, ten α-helices, six characteristic β–α–β motifs, and two disulfide cysteine bridges between 58–113Cys and 209–238Cys that may be responsible for the activity of enzyme.
3.2. Protein ionization and pKa measurements
The total titration curves of chitinase II were obtained by calculating the average net charge at each pH value (Figure (a)) using DS 4.0. The net charge and isoelectric pH (pI) of a protein depend on the content of ionizable groups and their pKa values. The maximum stability of chitinase II using CHARMm Polar H force fields was obtained at pH ranging from 2.2 to 3.8, respectively (Figure (b)) and the corresponding isoelectric points from 4.35 to 4.87, respectively (Table ). The pH-dependent relative folding energy was calculated using various modules present in DS (Spassov & Yan, Citation2008). The pH dependence of relative folding energy is calculated using the simplest null model of unfolded state i.e. ignoring the possible effects of any resting electrostatic interactions in unfolded state. In most cases, the results show a reasonable estimation of the pH stability which is comparable to the results obtained from more realistic models of the electrostatic interactions in the unfolded state. The total charge at a particular pH was lower than those computed using CHARMm Polar H force fields, while the relative folding energies were lower than those computed using CHARMm force fields. No significant changes were observed for both the models of chitinase II in the plot of total charge vs. pH. This suggests that the amino acids present in the signal peptide regions of chitinase II have little or no influence on the pH. Electrostatic interactions are widely believed to be primary factors relating to the pH-dependent phenomena. Since the charges carried by proteins are pH-dependent, the electrostatic interactions between residues of the proteins would be modulated by pHs. The electrostatic energy of chitinase II was found to be less at lower pHs, indicating that the chitinase II are stable at low pH. The pdb2gmx module of GROMACS generated the desirable protonation states by altering the configuration of the amino acids. Within the protein, the respective titratable residues were selected on the basis of information obtained from titration analysis. It was observed that chitinase II attained a more stable conformation at a lower pH over the 20 ns simulations.
Figure 1. The pH dependence of (a) total charge, (b) Relative Folding energy, at different protonated conditions of chitinase II. B1 and B2 represent chitinase II protein with CHARMm and CHARMm Polar H force fields, respectively, while B3 and B4 represent chitinase II without signal peptide typed with CHARMm and CHARMm Polar H force fields, respectively.
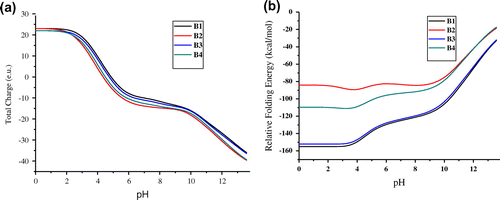
Table 1. Folding energy, Isoelectric point and electrostatic energy of chitinase II obtained using CHARMm and CHARMm Polar H force-fields
3.3. pH effect on the overall structure
The purpose of the energy value calculation was to ensure the stability of the protein during the MD simulations at a constant pH. The constant average fluctuation of temperature around 300 K suggested stable and accurate nature of the MD simulations performed. The results suggest that chitinase II showed a low value of average total energy of -1452083 kJ/mol at pH 5.
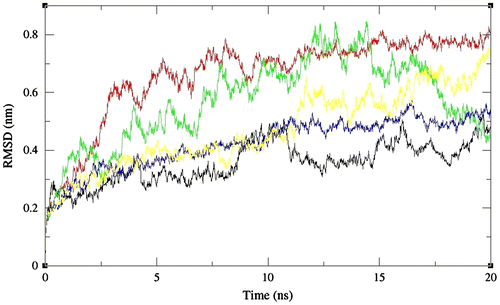
By considering the radius of gyration (Rg), we can conclude that chitinase II showed fluctuations throughout the simulations with the most compact conformations adopted after 10 ns at pH 3 (Figure ). The average RMSD values for chitinase II were found to be in the range of 0.35–0.66 nm indicating the prominent effect of pH on the structure, at pH 2 and 5 (Figure ).
Figure 2. The radius of gyration values as a function of time for full-length chitinase II obtained at pH 2 (black), 3 (red), 4 (green), 5 (blue), and 6 (yellow), respectively. Values were calculated with the use of Cα atoms.
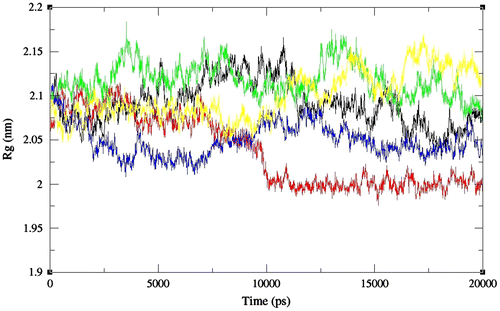
Figure 3. RMS deviation values as a function of time for full-length chitinase II obtained at pH 2 (black), 3 (red), 4 (green), 5 (blue), and 6 (yellow), respectively. Values were calculated with the use of Cα atoms.
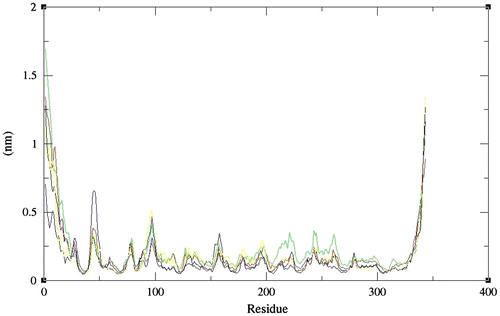
In addition to identify the more flexible regions, the RMSFs with reference to the residues were calculated for the protonated enzymes during simulations. The RMS fluctuations for chitinase II were comparable at all pHs except 4 indicating its activity for wider ranges of pH (Figure ).
Figure 4. Average RMS deviation values as a function of amino acid sequence numbers for full length chitinase II obtained at pH 2 (black), 3 (red), 4 (green), 5 (blue), and 6 (yellow), respectively. Values were calculated with the use of Cα atoms.
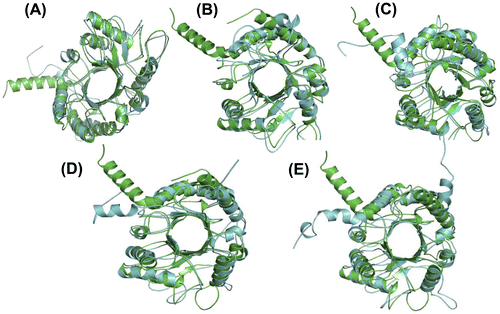
To establish the conformational changes, the average structures at each pH were calculated. The TIM-Barrel conformation of chitinase II was maintained during the 20 ns timescales of simulations. The RMS deviations of native and simulated structures of chitinase II were found to be 2.029, 1.924, 2.086, 1.708, and 2.108 Å at pH 2–6, respectively. The lowest RMS deviations for chitinase II were found at pH 3 and pH 5 (Figure ).
Figure 5. The superimposition of native and simulated structures of full-length chitinase II at 300 K, pH 2–6 from (A) to (E), respectively. Green and Blue represent native and the simulated chitinase II structures, respectively. Thickness of the coil represents the fluctuations of heavy atoms around the average structure. The RMS deviations were found to be 2.029, 1.924, 2.086, 1.708, and 2.108 Å at pH 2–6, respectively.
4. Conclusions
The structural effects arising from pH-induced changes on chitinase II enzymes have been performed using MD simulations method. pH is considered to be a major factor in solvent medium for the proper functioning of enzymes. We presented a titration curve and pK calculations of the titratable sites at a particular pH. Our results suggested that the maximum stability and optimal activity of chitnase II favor an acidic pH. The chitinase II remained in their close TIM barrel conformations during the MD simulations. Moreover, it was useful to study the deviations of secondary structure elements present in chitinase II at various pHs.
| Abbreviations | ||
| SSBP | = | Suren Singh Bernard Prior |
| RMSD | = | Root Mean Square Deviation |
| MD | = | Molecular dynamics |
| GROMACS | = | GROningen MAchine for Chemical Simulations |
| OPLS-AA/L | = | Optimized Potential for Liquid Simulations/all atoms |
Acknowledgments
The authors would like to express their acknowledgment to the CHPC South Africa, and Henan University of Technology, China.
Additional information
Funding
Notes on contributors
Faez Iqbal Khan
Faez Iqbal Khan received the PhD degree in Computational Chemistry and Bioinformatics from Durban University of Technology, South Africa in 2015. He got his BSc and MSc degrees in Biomedical Science and Bioinformatics from University of Delhi and Jamia Millia Islamia, respectively. He is currently a postdoctoral researcher at Henan University of Technology, China, in the field of Computational Science associated with Biomolecules, mainly focuses on structural and dynamics behavior of protein and enzymes.
References
- Anwer, K., Sonani, R., Madamwar, D., Singh, P., Khan, F., Bisetty, K., ... Hassan, M. I. (2015). Role of N-terminal residues on folding and stability of C-phycoerythrin: Simulation and urea-induced denaturation studies. Journal of Biomolecular Structure and Dynamics, 33, 121–133.10.1080/07391102.2013.855144
- Anwer, K., Rahman, S., Sonani, R. R., Khan, F. I., Islam, A., Madamwar, D., ... Hassan, M. I. (2016). Probing pH sensitivity of αC-phycoerythrin and its natural truncant: A comparative study. International Journal of Biological Macromolecules, 86, 18–27.10.1016/j.ijbiomac.2016.01.046
- Berardi, M. J., Shih, W. M., Harrison, S. C., & Chou, J. J. (2011). Mitochondrial uncoupling protein 2 structure determined by NMR molecular fragment searching. Nature, 476, 109–113.10.1038/nature10257
- Di Giambattista, R., Federici, F., Petruccioli, M., & Fenice, M. (2001). The chitinolytic activity of Penicillium janthinellum P9: Purification, partial characterization and potential applications. Journal of Applied Microbiology, 91, 498–505.10.1046/j.1365-2672.2001.01409.x
- Eswar, N., Eramian, D., Webb, B., Shen, M. Y., & Sali, A. (2008). Protein structure modeling with MODELLER. Methods in Molecular Biology, 426, 145–159.10.1007/978-1-60327-058-8
- Flanagan, M. A., Ackers, G. K., Matthew, J. B., Hanania, G. I., & Gurd, F. R. (1981). Electrostatic contributions to the energetics of dimer-tetramer assembly in human hemoglobin: pH dependence and effect of specifically bound chloride ions. Biochemistry, 20, 7439–7449.10.1021/bi00529a018
- Gramany, V., Khan, F. I., Govender, A., Bisetty, K., Singh, S., & Permaul, K. (2015). Cloning, expression, and molecular dynamics simulations of a xylosidase obtained from Thermomyces lanuginosus. Journal of Biomolecular Structure and Dynamics, 1–12.10.1080/07391102.2015.1089186
- Hamid, R., Khan, M. A., Ahmad, M., Ahmad, M. M., Abdin, M. Z., Musarrat, J., & Javed, S. (2013). Chitinases: An update. Journal of Pharmacy And Bioallied Sciences, 5, 21–29.
- Henrissat, B. (1999). Classification of chitinases modules. EXS, 87, 137–156.
- Herrera-Estrella, A., & Chet, I. (1999). Chitinases in biological control. EXS, 87, 171–184.
- Horng, J. C., Tracz, S. M., Lumb, K. J., & Raleigh, D. P. (2005). Slow folding of a three-helix protein via a compact intermediate †. Biochemistry, 44, 627–634.10.1021/bi048852p
- Humphrey, W., Dalke, A., & Schulten, K. (1996). VMD: Visual molecular dynamics. Journal of Molecular Graphics, 14, 27–28.
- Khan, F. I., Bisetty, K., Singh, S., Permaul, K., & Hassan, M. I. (2015). Chitinase from Thermomyces lanuginosus SSBP and its biotechnological applications. Extremophiles, 19, 1055–1066.10.1007/s00792-015-0792-8
- Khan, F. I., Govender, A., Permaul, K., Singh, S., & Bisetty, K. (2015). Thermostable chitinase II from Thermomyces lanuginosus SSBP: Cloning, structure prediction and molecular dynamics simulations. Journal of Theoretical Biology, 374, 107–114.10.1016/j.jtbi.2015.03.035
- Khan, F. I., Aamir, M., Wei, D. Q., Ahmad, F., & Hassan, M. I. (2016). Molecular mechanism of Ras-related protein Rab-5A and effect of mutations in the catalytically active phosphate-binding loop. Journal of Biomolecular Structure and Dynamics, 1–36.10.1080/07391102.2015.1134346
- Khan, F. I., Shahbaaz, M., Bisetty, K., Waheed, A., Sly, W. S., Ahmad, F., & Hassan, M. I. (2016). Large scale analysis of the mutational landscape in β-glucuronidase: A major player of mucopolysaccharidosis type VII. Gene, 576, 36–44.10.1016/j.gene.2015.09.062
- Khan, F. I., Wei, D. Q., Gu, K. R., Hassan, M. I., & Tabrez, S. (2016). Current updates on computer aided protein modeling and designing. International Journal of Biological Macromolecules, 85, 48–62.10.1016/j.ijbiomac.2015.12.072
- Kuzmanic, A., & Zagrovic, B. (2010). Determination of ensemble-average pairwise root mean-square deviation from experimental B-factors. Biophysical Journal, 98, 861–871.10.1016/j.bpj.2009.11.011
- Lopes, P. E., Huang, J., Shim, J., Luo, Y., Li, H., Roux, B., & MacKerell, A. D. Jr. (2013). Polarizable force field for peptides and proteins based on the classical drude oscillator. Journal of Chemical Theory and Computation, 9, 5430–5449.10.1021/ct400781b
- Maheshwari, R., Bharadwaj, G., & Bhat, M. K. (2000). Thermophilic fungi: Their physiology and enzymes. Microbiology and Molecular Biology Reviews, 64, 461–488.10.1128/MMBR.64.3.461-488.2000
- Matthew, J. B., Gurd, F. R., Garcia-Moreno, B., Flanagan, M. A., March, K. L., & Shire, S. J. (1985). pH-dependent processes in protein. Critical Reviews in Biochemistry, 18, 91–197.10.3109/10409238509085133
- Nielsen, J. E., & McCammon, J. A. (2003). Calculating pKa values in enzyme active sites. Protein Science, 12, 1894–1901.10.1110/(ISSN)1469-896X
- OuYang, B., Xie, S., Berardi, M. J., Zhao, X., Dev, J., Yu, W., ... Chou, J. J. (2013). Unusual architecture of the p7 channel from hepatitis C virus. Nature, 498, 521–525.10.1038/nature12283
- Singh, S., Madlala, A. M., & Prior, B. A. (2003). Thermomyces lanuginosus: Properties of strains and their hemicellulases. FEMS Microbiology Reviews, 27, 3–16.10.1016/S0168-6445(03)00018-4
- Song, Y., Mao, J., & Gunner, M. R. (2009). MCCE2: improving protein pKa calculations with extensive side chain rotamer sampling. Journal of Computational Chemistry, 30, 2231–2247.
- Spassov, V. Z., & Yan, L. (2008). A fast and accurate computational approach to protein ionization. Protein Science, 17, 1955–1970.10.1110/(ISSN)1469-896X
- Stephens, D. E., Khan, F. I., Singh, P., Bisetty, K., Singh, S., & Permaul, K. (2014). Creation of thermostable and alkaline stable xylanase variants by DNA shuffling. Journal of Biotechnology, 187, 139–146.10.1016/j.jbiotec.2014.07.446
- Tran, H. T., Barnich, N., & Mizoguchi, E. (2011). Potential role of chitinases and chitin-binding proteins in host-microbial interactions during the development of intestinal inflammation. Histology and Histopathology, 26, 1453–1464.
- Vaaje-Kolstad, G., Houston, D. R., Riemen, A. H., Eijsink, V. G., & van Aalten, D. M. (2005). Crystal structure and binding properties of the Serratia marcescens chitin-binding protein CBP21. Journal of Biological Chemistry, 280, 11313–11319.10.1074/jbc.M407175200
- Van Der Spoel, D., Lindahl, E., Hess, B., Groenhof, G., Mark, A. E., & Berendsen, H. J. (2005). GROMACS: Fast, flexible, and free. Journal of Computational Chemistry, 26, 1701–1718.10.1002/(ISSN)1096-987X
- Yang, A. S., & Honig, B. (1993). On the pH dependence of protein stability. Journal of Molecular Biology, 231, 459–474.10.1006/jmbi.1993.1294