
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Secondary structures (SS) of proteins are of great importance to structural, molecular, and computational biology and chemistry. Accurate and reliable method for automatic SS assignment when only coarse-grained (CG) information is available is needed. RaFoSA, a novel, accurate, and reliable method for automatic SS assignment based on coordinates of alpha carbon (CAC) atoms alone is presented here. Results from RaFoSa have been rigorously compared to those from Dictionary of Protein SS (DSSP, the acclaimed gold-standard for automatic SS assignment) and STRIDE. Requiring only CAC, RaFoSA achieves an agreement of 96% (and 94%) with DSSP (and STRIDE) that require all-atom and hydrogen-bonding information. No known automatic SS assignment method based on CG system has ever achieved such agreement with DSSP and STRIDE. Furthermore, RaFoSA has been applied to a real-life problem and its possible use for ranking proteins in their order of SS-based stability is shown in this paper. Overall, RaFoSA’s abilities to accurately and reliably assign SS to CG or all-atom protein systems make this work important. Furthermore, it must be emphasized that SS assignment by RaFoSA is different from (and is more rigorous than) SS prediction from amino acids sequence. Indeed, SS assignment by RaFoSA can differentiate between frames from molecular dynamics simulations trajectories, while existing methods for SS prediction from amino acid sequence cannot. Source codes and a webserver implementation of RaFoSA are available at http://bioinformatics.center/RaFoSA.
Public Interest Statement
Secondary structures (SS) of proteins are of great importance to structural, molecular, and computational biologists and chemists. Therefore, there is a need to develop accurate and reliable method for SS assignment when only alpha carbon information is available. This is why RaFoSA, a novel and reliable method for automatic SS assignment based on the coordinates of alpha carbon (CAC) atoms alone has been developed and presented here. RaFoSA accurately and reliably assigns SS to amino acids of proteins and achieves an agreement of 96% (and 94%) with DSSP (and STRIDE) that require all-atom and hydrogen-bonding information. RaFoSA may also help in ranking proteins based on the stability of their secondary structures. Source codes of RaFoSA are available at http://bioinformatics.center/tools/rafosa. A webserver that implements RaFoSA is available at http://bioinformatics.center/servers/rafosa.
Competing Interests
The author declare no competing interest.
1. Introduction
There are many important purposes (Cabaleiro-Lago, Szczepankiewicz, & Linse, Citation2012; Cino, Choy, & Karttunen, Citation2012; Emr & Silhavy, Citation1983; Ji & Li, Citation2010; Konvalinová et al., Citation2015; Myers & Oas, Citation2001) for which structural, molecular, and computational biologists (SMCB) need SS of proteins even when only coarse-grained (CG) information of the protein is available. Proper visualization of the structure and dynamics of proteins following CG molecular dynamics simulations (which continue to gain popularity) is one of the most notable of such purposes (Humphrey, Dalke, & Schulten, Citation1996). Without SS information, the molecular systems (MS) of interest are more difficult to study and less appealing to sight (Figure ). Availability of SS information removes the ordeal and allows proper visualization and smoother study of the static and dynamic properties of the MS of interest. In addition, SS changes of MS are frequently used to support evidences for, or against, structural stabilities of MS (Camilloni, De Simone, Vranken, & Vendruscolo, Citation2012; Pires, Ascher, & Blundell, Citation2014; Provencher & Gloeckner, Citation1981).
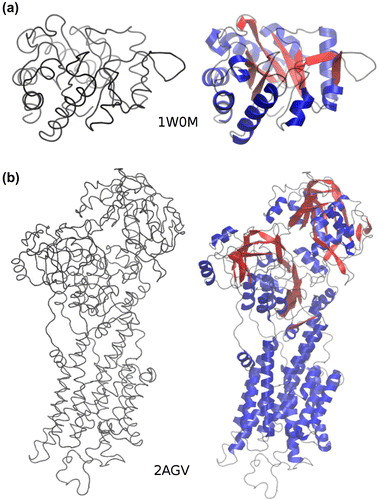
Figure 1. RaFoSA improves visualization of molecules when only coarse-grained information is available. (a) 1W0M, and (b) 2AGV are protein molecules arranged in an apparent order of increasing SS complexities. The visualizations on the left side (when all-atom and SS information are lacking) are not helpful and do not allow one to clearly differentiate (more structurally stable) sheets from (less structurally stable) coils. In contrast, the visualizations on the right side when RaFoSA provides SS information are more useful/helpful and are visually appealing. With the SS assigned, one can see the residues that form more structurally stable sheets (red) and helixes (blue), or less structurally stable coils (black).
Using random forests classification (Breiman, Citation2001), I have developed a computer program (RaFoSA) and a webserver for automatic SS assignment. RaFoSA (which is freely available at http://bioinformatics.center/RaFoSA) requires only the coordinates of alpha carbon atoms (CAC). Therefore, it works for both CG and all-atom protein systems (i.e. regardless of whether all-atom information is available or not). Rather than manually looking for “human rules” (i.e. manual deterministic approach) that could guide SS assignment, the computer was allowed to learn the SS classification of proteins based on random forests algorithm (Breiman, Citation2001).
Direct and automatic SS assignment by RaFoSA is straightforward, and better than using a pipeline that uses a prediction approach to first reconstruct the all-atom details of the protein (Krivov, Shapovalov, & Dunbrack, Citation2009) and then uses the predicted all-atom structure to predict SS. Unlike RaFoSA, such pipeline would be slow and vulnerable to errors propagation and compounding errors.
Although it is shown in this paper that RaFoSA requires only CAC and achieves 96% (and 94%) agreements with DSSP (and STRIDE)—the acclaimed gold-standard for automatic SS assignment—it is important to mention that other SS assignment methods based on CAC exist in the literature. Nonetheless, RaFoSA has better accuracy (and agreement with the gold-standard) than all other methods (Table ) such as P-SEA (Labesse, Colloc’h, Pothier, & Mornon, Citation1997), VoTAP (Dupuis, Sadoc, & Mornon, Citation2004), SEGNO (Cubellis, Cailliez, & Lovell, Citation2005), KAKSI (Martin et al., Citation2005), P-Curve (Sklenar, Citation1989), and DEFINE (Richards & Kundrot, Citation1988). A review of the SS assignment algorithms for these methods has been published elsewhere (Andersen & Rost, Citation2009).
Table 1. Agreements between various SS assignment methods and DSSP (Kabsch & Sander, Citation1983) and STRIDE (Heinig & Frishman, Citation2004)
2. System and methods
2.1 Data-set and machine learning
3D structures of non-homologous protein molecules (with less than 30% sequence similarity) were obtained from Brookhaven Protein Data Bank (Berman et al., Citation2000), and energy-minimized using AMBER (Salomon-Ferrer, Case, & Walker, Citation2013) force-fields (ff12SB). “Coordinates of alpha carbon (CA) atoms” (CAC) were extracted from the energy-minimized structures. For each residue in a given protein, 30 CAC-based features (Figure , Supplementary Table S1) were extracted and used for training random forests (RFC) (Breiman, Citation2001) machine-learning classifiers. Target labels were DSSP-assigned (Andersen, Palmer, Brunak, & Rost, Citation2002; Kabsch & Sander, Citation1983) SS classes. Eight RFC, one RFC for each of the seven SS classes and one for unknown class, were trained. With multidimensional grid search in Scikit-learn (Pedregosa et al., Citation2011), parameter spaces were explored using 10-fold cross-validation. The optimal model parameters found are summarised in the README file in RaFoSA’s source code downloadable from RaFoSA’s webpage.
Figure 2. Features used in RaFoSA. One of the features is the residue type (which is any of the 20 standard amino acids or “X” for any non-standard amino acid). Other features are related to alpha carbon (CA) atoms. Six of the features are CA-CA distances (a), such as di−1,i+1. Other six of the features are CA-CA-CA angles (b), such as ai−2,i,i+2. Four of the features are sign or angle of CA-CA-CA-CA torsional angles (c), such as ti−1,i,i+1,i+2. While the remaining features are based on residue–residue contacts (c), such as Ci,4.0. “i” is the index of the current residue. “i − 1” (or “i + 1”) is the index of the residue immediately before (or immediately after) the current residue.
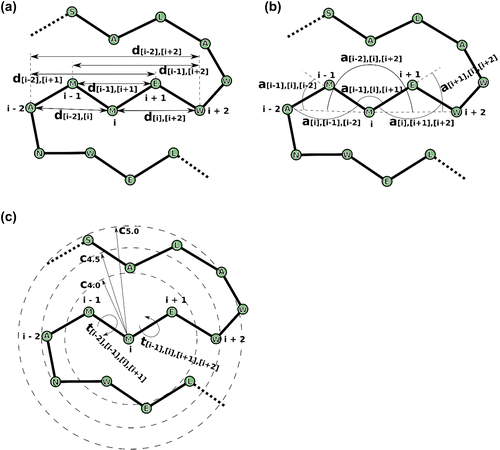
The features (i.e. variables, Figure ) used for the machine learning models are able to describe the local conformation and geometry of every five consecutive amino acids −i−2, i−1, i, i+1, and i+2 (Figure ). Such conformation and geometry are able to capture the secondary structures (SS) of proteins.
At this point, it is important to give the basis of random forests classification. A random forest classifier is an ensemble learning method that uses various sub-samples of the data-set to fit a predefined number of classifiers for the classification task of interest based on decision trees. Together, the decision trees make up the forest called the random forest classifier. When a trained random forest classifier is used for classification, each of the decision trees that make up the forest is used to predict the class of the current sample. The mode of the classes predicted by all decision trees is reported as the class for the current sample in the classification problem. It has been shown that random forest classification (as well as the analogous random forest regression) has many advantages (Amaratunga, Cabrera, & Lee, Citation2008; Biau, Citation2012; Breiman, Citation2001). Notably, with random forest, over-fitting is not an issue (Amaratunga et al., Citation2008; Biau, Citation2012; Breiman, Citation2001). It is very fast and its accuracy is overall better than those of other current machine learning algorithms (Amaratunga et al., Citation2008; Biau, Citation2012; Breiman, Citation2001).
2.2 Secondary structure assignment
Each trained RFC predicts the assignment of the current residue to the model’s SS class. For example, RFC for alpha helix (H) predicts whether the current residue should be assigned H or not; and so on. In a situation where each of two or more models reports a “true” for the current residue, “HBEGITS” order of preference is followed such that S is the least preferred. This order generally corresponds to decreasing false positivity rate and is consistent with DSSP’s (Carter, Andersen & Rost, Citation2003; Kabsch & Sander, Citation1983) approach. After SS had been assigned to all residues, all the assignments are scanned for consistency. It is ensured that whenever any of H, G, or I is assigned, it is assigned to three or more consecutive residues, because a reasonable helical fragment/structure should be made up by at least three residues. This is done by either extending or removing the H, G, or I segments based on the probabilities reported by the random forest classifiers for each of the amino acid positions. In a similar way, whenever B or E is assigned, it is assigned to two or more consecutive residues. This is similar to the approach DSSP (Carter, Andersen, & Rost, Citation2003; Kabsch & Sander, Citation1983) and other SS assignment methods (Andersen & Rost, Citation2003; Martin et al., Citation2005) use.
2.3 Coarse-grained MD simulations
Using Gō (Taketomi, Ueda, & Gō, Citation1975) model implemented in Cafemol (Kenzaki et al., Citation2011), coarse-grained molecular dynamics (MD) simulations of wild type Bacillus subtilis LipA (PDB code: 2QXU), and its variant X mutant (PDB code: 3QZU) were carried out at three different temperatures (300 K, 350 K, and 400 K) and two replicates each. Langevin dynamics was used. Each simulation was at 40 femtoseconds time-step and 2 microseconds long.
2.4 Assessment of stability of proteins
An empirical measure for quantifying the stability of a protein molecule (SS-stability score, SSS) based on its secondary structure constituents was developed. Sheets are regarded as the most stable (and have a weight of 1.5), followed by helixes (with a weight of 1.0), while coils are the least stable (with a weight of 0.0). The overall SSS of a protein is a weighted sum of the number of residues assigned to sheets or helixes divided by fifth-sixth of its number of residues . From the foregoing, a hypothetical protein that is made up only by coils is unstable (and has an overall SSS of 0.0). And the more the number of amino acids in sheets (as well as in helixes) a protein has, the higher its SSS. For purpose of illustrating the concept, I used the developed RaFoSA and SSS (derived from RaFoSA-assigned SS) to assess the SSS of wild type B. subtilis LipA, and its variant X mutant (Augustyniak et al., Citation2012). The findings are presented in the results section.
3. Results and discussion
3.1 Accuracy and reliability of RaFoSa
RaFoSA was rigorously tested on 1,000 (randomly selected, independent) protein molecules from the Protein Data Bank (Berman et al., Citation2000). See Supplementary Table S2 for the list of the protein molecules used. Despite working with only CAC, RaFoSA’s SS assignments agree with those of DSSP (Andersen et al., Citation2002; Carter et al., Citation2003; Kabsch & Sander, Citation1983), STRIDE (Heinig & Frishman, Citation2004), and PSEA (Labesse et al., Citation1997). RaFoSA achieves an agreement of 96.2% (and 93.9%) with DSSP (and STRIDE) that require all-atom and hydrogen-bonding information, which are not always available. Comparison of PSEA and STRIDE (which requires all-atom details) to DSSP shows only 82% and 95.9% agreements, respectively. Such relatively low agreements between PSEA and DSSP, and between STRIDE and DSSP are already known (Klose, Wallace, & Janes, Citation2010; Labesse et al., Citation1997; Martin et al., Citation2005; Zhang, Dunker, & Zhou, Citation2008). Indeed, these make PSEA and STRIDE less desirable alternatives to RaFoSA (that is presented here), even if all-atom information is available. Furthermore, RaFoSA is able to assign SS to each of the amino acids in any given protein whatsoever, using either seven-class system or three-class system (see the next paragraph) even for CG protein models that have only CA atoms. In addition, comparison of RaFoSA to VoTAP (Dupuis et al., Citation2004), SEGNO (Cubellis et al., Citation2005), KAKSI (Martin et al., Citation2005), P-Curve (Sklenar, Citation1989), and DEFINE (Richards & Kundrot, Citation1988) show RaFoSA to be superior (Table ).
The proportion of 238,216 residues (from the 1,000 proteins used for RaFoSA’s evaluation) assigned to alpha helix (H), beta sheet (B), strand (E), 3–10 helix (G), pi-helix (I), turn (T), and coil/bend (S) are presented in Figure (a), and are compared therein to those from DSSP and STRIDE. Based on this seven-class SS information, RaFoSA and DSSP agree by 93.0%, while RaFoSA and STRIDE agree by approximately 80.0%. It must be noted that PSEA does not use seven-class system, therefore it is not included in the comparison shown in Figure (a).
Figure. 3. Agreement between RaFoSA’s SS assignment and SS assignment by some existing methods. (a) Proportion of the assigned SS that falls in each of the seven SS classes–alpha helix (H), beta sheet (B), strand (E), 3–10 helix (G), pi-helix (I), turn (T), and coil/bend (S). (b) Proportion of the assigned SS that falls within each of the three SS classes–sheet, s; helix, h; coil, c–based on mapping 1, M1 (“HBEGITS” → “hcscccc”). (c) Same as b, but based on mapping 2, M2 (“HBEGITS” → “hsshhcc”). Panels d to f (based on M1) and panels g to h (based on M2) show the degree of agreement between RaFoSA and each of the other methods. Using M1, agreements between SS assignments by RaFoSA and DSSP are shown in d, between RaFoSA and STRIDE are shown in e, and between RaFoSA and PSEA are shown in f. Similar information are shown in g, h, and i, but based on M2. The columns in each of the matrixes are for RaFoSA-assigned Sheets, Helixes, and Coils, respectively. The rows are for the other SS methods RaFoSA is compared to. The intensity of the blue color in the leading diagonal of each of the matrixes show the degree of agreement between RaFoSA and the other SS assignment method. Number of amino acids per SS are shown in j (sheet), k (helix), and l (coil).
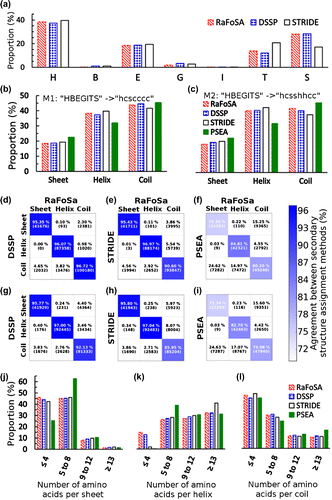
The seven-class SS information is often converted to three-class SS information (sheet, s; helix, h; and coil, c) for simplicity and when being used by molecular visualization software systems (Humphrey et al., Citation1996) or whenever it is used for assessing stability of MS (Camilloni et al., Citation2012). The seven-class SS was therefore converted into its three-class form using the two most commonly used (Andersen & Rost, Citation2003; Andersen et al., Citation2002; Carter et al., Citation2003; Heinig & Frishman, Citation2004; Kabsch & Sander, Citation1983) mappings—Mapping 1 (M1): “HBEGITS” → “hcscccc”; Mapping 2 (M2): “HBEGITS” → “hsshhcc”. The proportions of the SS assigned to sheet (s), helix (h), and coil (c) by RaFoSA, DSSP, STRIDE, and PSEA are shown in Figure (b) (based on M1) and Figure (c) (based on M2). RaFoSA agrees well with DSSP and STRIDE.
The agreements were further assessed on residue-by-residue basis using both M1 and M2. A hypothetical situation in which two methods agree perfectly will have the leading diagonal of the heatmaps (shown in Figure (d) to Figure (i)) to be deep blue (100% agreement) and the off-diagonal to be plain white. Based on M1, RaFoSA and DSSP (Figure (d)) agree in 96.2% of all their SS assignments (95.4% for sheets, 96.1% for helix, and 96.7% for coils). RaFoSA and STRIDE (Figure (e)) agree in 93.9% of all their SS assignments, while RaFoSA and PSEA (Figure (f)) agree in 80.8% of all their SS assignments. Based on M2, RaFoSA and DSSP (Figure (g)) agree in 94.8% of all their SS assignments, RaFoSA and STRIDE (Figure (h)) agree in 92.2% of all their SS assignments, while RaFoSA and PSEA (Figure (i)) agree in 80.0% of all their SS assignments.
For further comparison, the distributions of the number of residues in each sheet (Figure (j)), helix (Figure (k)), and coil (Figure (l)) were calculated. In all, RaFoSA shows great agreement with existing SS assignment methods, and (more importantly) works when existing gold-standard method, DSSP, cannot (namely when all-atom information is not available).
3.2 RaFoSA identifies stable variant of proteins
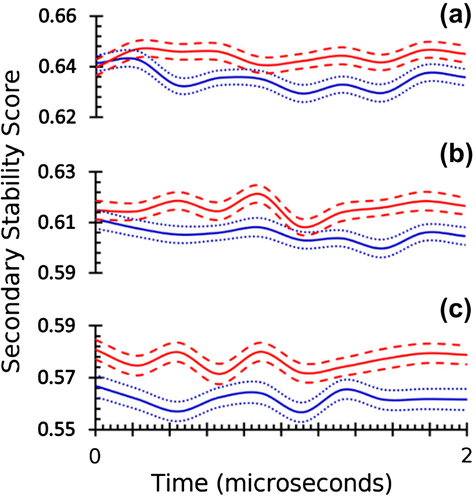
Since RaFoSA is accurate at, and reliable for, SS assignment regardless of whether all-atom information is available or not, RaFoSA was used to assign SS to each of the 12,000 frames (1,000 frames × 2 molecules × 2 replicates × 3 temperatures) from 24 microseconds coarse-grained (CG) molecular dynamics (MD) simulations of wild type B. subtilis LipA (WTLA), and its variant X mutant (XMLA). The secondary stability score (SSS, see methods section) computed from RaFoSA-assigned SS clearly identifies XMLA as being more stable than WTLA (Figure ). This agrees with previous experiments (Augustyniak et al., Citation2012), and provides additional evidence that supports the accuracy and reliability of RaFoSA as well as suggests additional potential application of RaFoSA. It must be noted that analysis of stability of protein fold and identification of SS associated with protein function can be done in many other ways, most of which are more rigorous than the quick SS-based presented used in this example.
Figure. 4. Structural stability score based on RaFoSA-assigned SS identifies protein’s structurally stable variant. Structural stability of wild type B. subtilis LipA (blue, 2QXU), and its variant X mutant (red, 3QZU) over the simulation time at three different temperatures—(a) 300 K, (b) 350 K, and (c) 400 K—are shown. The solid lines are the means, while the dotted/dashed lines are one standard deviation above or below the respective mean values.
3.3 Source codes and webserver of RaFoSA
Source codes of RaFoSA are available at http://bioinformatics.center/tools/rafosa for free download. Once the user had downloaded and extracted RaFoSA into the desired directory, he/she can execute “python full/path/to/RaFoSA.py full/path/to/pdbFile.pdb” to assign SS to the amino acids of the protein contained in the specified pdb file. This is supported on all major operating systems. A working Python programing language environment (with NumPy, SciKitLearn, and BioPython) is required. Additional information on how to set up RaFoSA, the format of RaFoSA’s output, and how to use RaFoSA with VMD are available on RaFoSA’s webpage.
Furthermore, a webserver (summarized in Figure ) that implements RaFoSA is made available at http://bioinformatics.center/servers/rafosa so as to further increase the user-friendliness of RaFoSA. The webserver supports all modern web browsers. User may provide input (Figure (a)) to the webserver in any of four ways: (1) by uploading PDB file, (2) by entering PDB content/format as text, (3) by specifying a valid PDB ID, or (4) by uploading trajectory as PSF and DCD files. Regardless of how the user submits the input, SS assignment is done for protein residues (i.e. for amino acids) alone, SS are assigned for all frames (Figure (b)) in the submitted data, and SS visualization (Figure (c)) is generated for the protein for each frame. Summary statistics for the assigned SS are also provided (see the line graph and doughnut chart in Figure (b) and Figure (c), respectively).
Figure. 5. RaFoSA webserver. The webserver accepts input (a) in any of four ways: (1) PDB file, (2) PDB content/format as text, (3) PDB ID, or (4) trajectory as PSF and DCD files. SS are assigned for all frames (b) in the submitted data, and SS visualization (c) is generated for the protein for each frame. Summary statistics (line graph, (b), and doughnut chart, (c)) are also provided for the assigned SS.

4. Conclusions
The need to be able to study biomolecules in silico at biologically meaningful timescale with limited available computation power has made coarse-grained (CG) molecular dynamics (MD) simulations to increasingly gain popularity and acceptance. However, “accurate and reliable method” for SS assignment to residues of protein molecules following their CG-MD simulations (or whenever all-atom information is lacking) does not exist. RaFoSA was developed to address this problem and to cater for this need. Therefore, RaFoSA is a new method (implemented as a software and as a webserver) for protein SS assignment when only CG information is available. This paper has shown evidences and confirmed that RaFoSA produces accurate and reliable results and works well even in situations where existing methods cannot work well. For example, DSSP (Andersen et al., Citation2002; Kabsch & Sander, Citation1983) and STRIDE (Heinig & Frishman, Citation2004) require all-atom information and hydrogen-bonding patterns. These make them to have very limited applications, to the extent that they are completely unusable when one is directly dealing with trajectories or snapshots from CG-MD simulations. On the other hand, RaFoSA works in all cases and produces accurate and reliable results that no other SS method (including CA-only methods such as PSEA) can match.
It must, at least, be noted that the method presented in this paper is not the same as SS prediction from sequence information that is already well-documented (Cuff & Barton, Citation2000; Kelley & Sternberg, Citation2009; Rost, Citation2001). More importantly, it must be noted that existing SS prediction methods that use sequence information fail, and cannot serve the purposes RaFoSA serves. However, this does not necessarily imply that SS prediction from sequence information is bad or unimportant. Nonetheless, it is important to emphasize that such SS prediction method cannot serve the extended purposes for which RaFoSA has been developed. For example, sequence-based SS prediction (SBSP) methods cannot appropriately assign SS to each residue in each frame of trajectories from CG-MD simulations the way RaFoSA does. Such SBSP methods would rather assign the same (and, therefore, incorrect) static SS (that would not change over the simulation trajectory) to each of the residues in each of the frames in the trajectories. Thus, unlike RaFoSA, such SBSP methods are unable to correctly capture the structural dynamical properties of proteins at all. On the other hand, RaFoSA can identify structurally stable variant of proteins based on SSS (such as in Figure ). SBSP methods (that are largely based on sequence homology) fail, because native SS compositions of two or more studied molecules (and thus their SSS) can be quite comparable and not differentiable in their native states, and at the beginning of the simulation (such as Figure (a) and Figure (b)). Therefore, only a method (such as RaFoSA) that can correctly assign SS to each frame in the trajectories could work for this purpose and similar purposes.
Anyone can obtain and use the source codes of non-commercial version of RaFoSA from http://bioinformatics.center/tools/rafosa. A webserver that implements RaFoSA for easy use is available at http://bioinformatics.center/servers/rafosa. User guides are also provided through the webpages.
Cover image
Source: Author
Supplementary material
The supplementary material for this paper is available online at http://dx.doi.10.1080/23312025.2016.1214061.
SupplementaryInformation_RaFoSA_revised_2.docx
Download MS Word (20.8 KB)Acknowledgements
I thank Dr Claus Andersen (for making a copy of DSSPCont available), Dr Gilles Labesse (for making a copy of PSEA available), and Dr David Case (for providing a free academic license of AMBER14). The extensive feedback from people (Dr Jiří Koubek, Chris YC Lo, ChiHong ChangChien, etc.) who have being using RaFoSA and/or offered criticisms that helped in improving RaFoSa and this paper are acknowledged.
Additional information
Funding
Notes on contributors
Emmanuel Oluwatobi Salawu
Emmanuel Oluwatobi Salawu received bachelor of technology (with Honors) in Physiology at Ladoke Akintola University of Technology, and later proceeded to study Computer Science at the University of Hertfordshire where he earned master of science (with Distinction). He is currently a Bioinformatics and Structural Biology PhD candidate at National Tsing Hua University. He has interests in molecular dynamics simulations, protein structures, machine learning, image analysis, epidemiology, and malaria research. The work presented here is part of his research activities involving secondary structures of proteins and machine learning.
References
- Amaratunga, D., Cabrera, J., & Lee, Y.-S. (2008). Enriched random forests. Bioinformatics, 24, 2010–2014.10.1093/bioinformatics/btn356
- Andersen, C. A. F., & Rost, B. (2003). Secondary structure assignment. Methods of Biochemical Analysis, 44, 341–363. Retrieved from http://doi.org/10.1002/0471721204.ch17
- Andersen, C. A. F., & Rost, B. (2009). Secondary structure assignment. Structural. Bioinformatics, 44, 459–484.
- Andersen, C. A. F., Palmer, A. G., Brunak, S., & Rost, B. (2002). Continuum secondary structure captures protein flexibility. Structure, 10, 175–184. Retrieved from http://doi.org/10.1016/S0969-2126(02)00700-1 10.1016/S0969-2126(02)00700-1
- Augustyniak, W., Brzezinska, A. A., Pijning, T., Wienk, H., Boelens, R., Dijkstra, B. W., … Reetz, M. T. (2012). Biophysical characterization of mutants of Bacillus subtilis lipase evolved for thermostability: Factors contributing to increased activity retention. Protein Science, 21, 487–497. Retrieved from http://doi.org/10.1002/pro.2031 10.1002/pro.2031
- Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissig, H., … Bourne, P. E. (2000). The protein data bank. Nucleic Acids Research, 28, 235–242. Retrieved from http://doi.org/10.1093/nar/28.1.235 10.1093/nar/28.1.235
- Biau, G. (2012, April). Analysis of a random forests model. Journal of Machine Learning Research, 13, 1063–1095.
- Breiman, L. (2001). Random forests. Machine Learning, 5–32. Retrived from http://doi.org/10.1023/A:1010933404324 10.1023/A:1010933404324
- Cabaleiro-Lago, C., Szczepankiewicz, O., & Linse, S. (2012). The effect of nanoparticles on amyloid aggregation depends on the protein stability and intrinsic aggregation rate. Langmuir, 28, 1852–1857. Retrieved from http://doi.org/10.1021/la203078w 10.1021/la203078w
- Camilloni, C., De Simone, A., Vranken, W. F., & Vendruscolo, M. (2012). Determination of secondary structure populations in disordered states of proteins using nuclear magnetic resonance chemical shifts. Biochemistry, 51, 2224–2231. Retrieved from http://doi.org/10.1021/bi3001825 10.1021/bi3001825
- Carter, P., Andersen, C. A., & Rost, B. (2003). DSSPcont: Continuous secondary structure assignments for proteins. Nucleic Acids Research, 31, 3293–3295.10.1093/nar/gkg626
- Cino, E. A., Choy, W. Y, & Karttunen, M. (2012). Comparison of secondary structure formation using 10 different force fields in microsecond molecular dynamics simulations. Journal of Chemical Theory and Computation, 8, 2725–2740. Retrieved from http://doi.org/10.1021/ct300323g 10.1021/ct300323g
- Cubellis, M. V., Cailliez, F., & Lovell, S. C. (2005). Secondary structure assignment that accurately reflects physical and evolutionary characteristics. BMC Bioinformatics, 6(4), 1.
- Cuff, J. A., & Barton, G. J. (2000). Application of multiple sequence alignment profiles to improve protein secondary structure prediction. Proteins: Structure, Function, and Genetics, 40, 502–511. Retrieved from http://doi.org/10.1002/1097-0134(20000815)40:3<502::AID-PROT170>3.0.CO;2-Q, 10.1002/(ISSN)1097-0134
- Dupuis, F., Sadoc, J.-F., & Mornon, J.-P. (2004). Protein secondary structure assignment through Voronoï tessellation. Proteins: Structure, Function, and Bioinformatics, 55, 519–528.10.1002/prot.10566
- Emr, S. D., & Silhavy, T. J. (1983). Importance of secondary structure in the signal sequence for protein secretion. Proceedings of the National Academy of Sciences, 80, 4599–4603. Retrieved from http://doi.org/10.1073/pnas.80.15.4599 10.1073/pnas.80.15.4599
- Heinig, M., & Frishman, D. (2004). STRIDE: A web server for secondary structure assignment from known atomic coordinates of proteins. Nucleic Acids Research, 32 (WEB SERVER ISS.), W500–W502.10.1093/nar/gkh429
- Humphrey, W., Dalke, A., & Schulten, K. (1996). VMD: Visual molecular dynamics. Journal of Molecular Graphics, 14, 33–38. Retrieved from http://doi.org/10.1016/0263-7855(96)00018-5xz, 10.1016/0263-7855(96)00018-5
- Ji, Y.-Y., & Li, Y.-Q. (2010). The role of secondary structure in protein structure selection. The European Physical Journal E, 32, 103–107.Retrieved from http://doi.org/10.1140/epje/i2010-10591-5 10.1140/epje/i2010-10591-5
- Kabsch, W., & Sander, C. (1983). Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers, 22, 2577–2637.Retrieved from http://doi.org/10.1002/bip.360221211, 10.1002/(ISSN)1097-0282
- Kelley, L. A., & Sternberg, M. J. E. (2009). Protein structure prediction on the web: A case study using the Phyre server. Nature Protocols, 4, 363–371.10.1038/nprot.2009.2
- Kenzaki, H., Koga, N., Hori, N., Kanada, R., Li, W., Okazaki, K. I., … Takada, S. (2011). Cafemol: A coarse-grained biomolecular simulator for simulating proteins at work. Journal of Chemical Theory and Computation, 7, 1979–1989. Retrieved from http://doi.org/10.1021/ct2001045 10.1021/ct2001045
- Klose, D. P., Wallace, B. A., & Janes, R. W. (2010). 2Struc: The secondary structure server. Bioinformatics, 26, 2624–2625.10.1093/bioinformatics/btq480
- Konvalinová, H., Dvořáková, Z., Renčiuk, D., Bednářová, K., Kejnovská, I., Trantírek, L., … Sagi, J. (2015). Diverse effects of naturally occurring base lesions on the structure and stability of the human telomere DNA quadruplex. Biochimie, 118, 15–25. Retrieved from http://doi.org/10.1016/j.biochi.2015.07.013 10.1016/j.biochi.2015.07.013
- Krivov, G. G., Shapovalov, M. V., & Dunbrack, R. L. (2009). Improved prediction of protein side-chain conformations with SCWRL4. Proteins: Structure, Function, and Bioinformatics, 77, 778–795.10.1002/prot.v77:4
- Labesse, G., Colloc’h, N., Pothier, J., & Mornon, J. P. (1997). P-SEA: A new efficient assignment of secondary structure from C alpha trace of proteins. Computer Applications in the Biosciences: CABIOS, 13, 291–295. Retrieved from http://doi.org/10.1093/bioinformatics/13.3.291
- Martin, J., Letellier, G., Marin, A., Taly, J.-F., de Brevern, A. G., & Gibrat, J.-F. (2005). Protein secondary structure assignment revisited: A detailed analysis of different assignment methods. BMC Structural Biology, 5, 17.10.1186/1472-6807-5-17
- Myers, J. K., & Oas, T. G. (2001). Preorganized secondary structure as an important determinant of fast protein folding. Nature Structural Biology, 8, 552–558. Retrieved from http://doi.org/10.1038/88626 10.1038/88626
- Pedregosa, F., Varoquaux, G., Gramfort, A., Michel, V., Thirion, B., Grisel, O., … Duchesnay, É. (2011, Oct). Scikit-learn : Machine learning in Python. Journal of Machine Learning Research, 12, 2825–2830.
- Pires, D. E. V., Ascher, D. B., & Blundell, T. L. (2014). DUET: A server for predicting effects of mutations on protein stability using an integrated computational approach. Nucleic Acids Research, 42, 1–6. Retrieved from http://doi.org/10.1093/nar/gku411
- Provencher, S. W., & Gloeckner, J. (1981). Estimation of globular protein secondary structure from circular dichroism. Biochemistry, 20, 33–37. Retrieved from http://doi.org/10.1021/bi00504a006 10.1021/bi00504a006
- Richards, F. M., & Kundrot, C. E. (1988). Identification of structural motifs from protein coordinate data: Secondary structure and first-level supersecondary structure. Proteins: Structure, Function, and Genetics, 3, 71–84.10.1002/(ISSN)1097-0134
- Rost, B. (2001). Review: Protein secondary structure prediction continues to rise. Journal of Structural Biology, 134, 204–218. Retrieved from http://doi.org/10.1006/jsbi.2000.4336 10.1006/jsbi.2001.4336
- Salomon-Ferrer, R., Case, D. A., & Walker, R. C. (2013). An overview of the Amber biomolecular simulation package. Wiley Interdisciplinary Reviews: Computational Molecular Science, 3, 198–210. Retrieved from http://doi.org/10.1002/wcms.1121
- Sklenar, H. (1989). Describing protein structure, 60, 46–60.
- Taketomi, H., Ueda, Y., & Gō, N. (1975). Studies on protein folding, unfolding and fluctuations by computer simulation. International Journal of Peptide and Protein Research, 7, 445–459.
- Zhang, W., Dunker, A. K., & Zhou, Y. (2008). Assessing secondary structure assignment of protein structures by using pairwise sequence-alignment benchmarks. Proteins: Structure, Function, and Bioinformatics, 71, 61–67.10.1002/(ISSN)1097-0134