
Abstract
Single nucleotide polymorphisms (SNPs) are common markers used for population genetics studies. Recently, copy number variations (CNVs) have been used to study genetic and phenotypic diversity within and among populations. Therefore, studies of the genetics of CNVs are important in the context of understanding evolutionary changes and genomic selection based upon genetic and phenotypic variation. The aim of this study was to analyse the distribution of the choline kinase beta (CHKB), Krüpple-like factor 6 (KLF6), glypican 1(GPC1) and cholinergic receptor muscarinic 3 (CHRM3) genes in five representative Chinese domestic yak breeds. The data generated by qPCR was transformed into log2 ratio and analysed using GraphPad (PRISM). The results show that the CNVs of CHKB, KLF6, and GPC1 genes presented more copy number losses in Tianzhu, Gannan and Plateau yak populations compared with the Datong and polled yak having relatively more copy number gains. However, the CHRM3 CNV showed more copy number gains in five yak populations. Therefore, these results indicate that there are relatively more copy number losses (deletion) in the yak populations; supporting the hypothesis that log2 ratio is more powerful at detecting loss than gain in copy number types. Taken together, these data provide information on the application genome CNVs in population genetics and suggest that the CNVs of the genes could exert a significant effect on phenotypic differences in yak populations.
PUBLIC INTEREST STATEMENT
Copy number variations (CNVs) are useful in the study of genetic and phenotypic variation among individuals. Population genetics of the CNVs are useful in the understanding of evolutionary changes and genomic selection within a given population. Yak (Bos grunniens) breeding is an economically important sector and supports millions of people for milk and beef produce. In a previous study, the CNVs of CHKB, KLF6, GPC1 and CHRM3 gene were shown to reside within copy number variable regions of yak populations and overlapped with quantitative trait loci (QTLs). In this report, copy number gain events may be due to artificial insemination, hybridization, and introgression of yak breeds. The study has indicated that copy number losses (deletion) are more abundant in yak populations. The results obtained will be an essential step towards understanding the molecular mechanisms underlying phenotypic variation among domesticated yaks in Qinghai-Tibet Plateau (QTP) of China.
1. Introduction
Morphological and molecular markers are often used to distinguish different mammalian species. However, characterization of the available genomic resources is significantly important in the study of genetic diversity and for selection of superior yak breeds and possibly other bovid species. Yaks (Bos grunniens) are of the same genus to the two domesticated cattle, Bos taurus and Bos indicus with chromosomal number of 60. The domestic yak (B. grunniens) is common in the larger and harshest highlands of the “the roof of the globe” Qinghai-Tibet Plateau (QTP) (Wiener, Han, & Long, Citation2003). The long hair and large horns are peculiar phenotypic characteristics of the yak breeds (Liu et al., Citation2013). Indeed the yak has become strongly integrated into Tibetans’ socio-cultural life (Liang et al., Citation2016), and it occupies a significant economic role in these mountainous regions of Asia (Medugorac et al., Citation2017); providing milk, meat, transportation (“boat of the plateau”), hair, draught power, and fuel for Tibetans and nomadic dwellers (Wiener, Han, & Long, Citation2003). Yaks have a long history of domestication and selective breeding (Wiener et al., Citation2003). Also, a unique physiological feature of yak breeds is their ability to survive extremely harsh environments at high altitudes, overcoming hypoxia, severely cold winters, and cool moist summers, and using limited grazing resources (Wiener et al., Citation2003). A major issue in the understanding of the mechanisms behind the development of the yak breeds is lack of genome DNA sequence information; therefore, a study elucidating the population genetics of copy number variations in yaks could lead to significant economic gains with respect to the breeding objectives. A population genetics survey based on CNVs has been reported for cattle breeds (Liu et al., Citation2010; Xu et al., Citation2016). Several studies have revealed that the copy number of variable regions in the genome can shape genetic networks and regulate gene expression, thereby contributing to inter-individual phenotypic variability (Conrad et al., Citation2010; Freeman et al., Citation2006). Notably, the CNVs encompassed more actual nucleotide content per genome compared to SNPs, further underscoring the importance of CNVs in the evolution of genetic diversity (Redon et al., Citation2006).
Therefore, the population genetics of CNVs is important in the understanding of evolutionary changes and the application of genomic selection based on genetic and phenotypic variation within a given population. Copy number variation contributes to the creation of novel genes which may allow genotypic and phenotypic diversification of the repertoire of such genes in the response to changing environments (Iskow, Gokcumen, & Lee, Citation2012). For instance, a study by Lee et al. (Citation2013) showed that there are a significant copy number variations present for the olfactory receptor (OR) genes in cattle, which is significant in determining individual and/or breed-specific differences in olfaction capacity. Human Amylase1 (AMY1) CNVs are thought to driven by natural selection and may have potential roles in driving evolutionary biological changes (Fernández & Wiley, Citation2017). For instance, AMY1 copy number is observed to be higher in populations that consume high-starch diets which are thought to have resulted from positive or directional selection and relatively lower numbers of copies are found in populations consuming lower starch diets due to genetic drift (Perry et al., Citation2007). Sjödin and Jakobsson (Citation2012) in their review suggested that recombination, mutation, selection, and demography are major forces in population genetics of CNVs. Evolutionary processes, domestication and artificial selection are thought to be the main factors responsible for genomic changes including CNVs and phenotypic divergence in animal productions (Kijas et al., Citation2009; Novembre & Ramachandran, Citation2011; Parsch & Ellegren, Citation2013). The study of CNVs has become important and they are an abundant source of genetic and phenotypic diversity (Liu et al., Citation2010), which is associated to complex disease and phenotypes (Stankiewicz & Lupski, Citation2010; Yang et al., Citation2007; Zhang, Youji, Yong, & Xingxu, Citation2015).
Genomic copy number variation of large segments DNA (at least 1 kb in size), are composed of both gains (insertions or duplications) and losses (deletions or null genotypes) in comparison with a designated reference genome sequence (Scherer et al., Citation2007). Genic CNV regions (CNVRs) have the potential to affect phenotypes variation, gene expression, and adaptation through the disruption of gene dosages (Weischenfeldt, Symmons, Spitz, & Korbel, Citation2013) and altering transcription levels of genes (Pollex & Hegele, Citation2007). CNVs have been well studied and linked to various phenotypic traits and diseases in humans and rodents (Almal & Padh, Citation2012; Cook & Scherer, Citation2008; Girirajan et al., Citation2013). The association between the CNVs and production traits of economic interest had been reported in swine (Chen et al., Citation2012). Iafrate et al. (Citation2004) revealed that copy number variations are an important genome resource in humans.
In the past, little research has been conducted on yak CNVs, focusing mostly on surveying CNVRs using array comparative genomic hybridization (aCGH) (Zhang, Jia, et al., Citation2014), BovineHD Genotyping Bead Chip Array (Quanwe et al., Citation2014) and next-generation sequencing (NGS) (Zhang et al., Citation2016). In recent years, relative copy number distributions were determined for the CNVs of myosin heavy chain 3 (MYH3), mitogen-activated protein kinase 10 (MAPK10) and leptin receptor (LEPR) genes and revealed considerable diversity in genetic copy numbers in Chinese cattle (Liu et al., Citation2016; Shi et al., Citation2015; Xu et al., Citation2013). Previous studies have using a read depth approach in the genome resequencing analysis of CNVs for choline kinase beta (CHKB), Krüpple-like factor 6 (KLF6), glypican 1(GPC1) and cholinergic receptor muscarinic 3 (CHRM3) genes has identified copy number variable regions in yak breeds (Zhang et al., Citation2016). Zhang et al. (Citation2016) also suggested that the CNVRs are an important genetic resource for phenotypic and genotypic variations for both yak and other bovid species, and these earlier studies have provided the raw materials for the current study. To date, there is no report made on the study on population genetics of copy number variations of the CHKB, KLF6, GPC1 and CHRM3 genes in Chinese domestic yak breeds by using real-time PCR technique.The main aim this study was to analyse copy number variations of the CHKB, KLF6, GPC1 and CHRM3 genes in Chinese domestic yak breeds. The results obtained from this work will be an essential step towards deciphering the molecular mechanisms underlying trait variation among the domesticated yaks in Qinghai-Tibet Plateau (QTP) of China.
2. Materials and methods
2.1. Study areas and sample collections
The main goal of Chinese domestic yak breeding comprises enhanced production of meat and milk, whilst maintaining and conserving genetic diversity of yak populations. Five representative Chinese domestic yak breeds were selected to study, namely, the polled yak (Datong Yak Farm in Qinghai Province, altitude 3200 m), Tianzhu white yak (Tianzhu Tibetan Autonomous County in Gansu Province, altitude 3000 m), plateau yak (northern and southern Qinghai Province, altitude 3700–4700 m), Datong yak (Datong Yak Farm in Qinghai Province, altitude 3200 m), and Gannan yak (Gansu Province, bordering Sichuan and Qinghai, altitude 3300–4400 m). The origins of the five breeds are shown in Figure .
Figure 1. Geographic map of the five yak breeds.
Fresh blood samples were collected from a total of 477 yaks (222 Datong yaks, 165 polled yaks, 30 Tianzhu yaks, 30 Gannan yaks, and 30 plateau yaks). The blood samples were taken from the jugular vein and collected in vacuum tubes (5 mL) containing anticoagulant citrate. All blood samples were stored at −80°C prior to DNA extraction. The animals were in good physical condition and assumed to not be genetically related. All yaks were grazing on natural pastures without feed supplementation, under similar feeding conditions and management.
2.2. Genomic DNA isolation
Genomic DNA (gDNA) was purified using clot blood DNA purification kit (CWBIO, China, Beijing) and cleaned using the genomic DNA clean up (>10kb) of E.Z.N.A. microelute DNA clean-up Kit (Omega Bio-tek, Georgia, USA) following the manufacturer’s instructions. The concentration and quality of the DNA was determined using a NanoDrop Biophotometer 2000 (Thermo Fisher Scientific Inc.) and through electrophoresis on 1% agarose gels and stained with ethidium bromide.
2.3. Primer design and PCR amplification
Using bovine genomic sequences, primers were designed for CNV of KLF6, CHKB, GPC1 and CHRM3 genes using the Primer v5.0 software (Shi et al., Citation2015) and the qPCR primers designed (HTTP://bitesizebio.com/10041/a-step-by-step-guide-to-designing-qPCR-primers/.) (Table ). The primers were designed so that the PCR product/amplicon size was <200bp (80–200 bp), melting temperature with ranges between 57 and 63°C (with a maximum difference of 3°C in the Tm’s of the two primers), length of primers (18–22 bp), GC content (50–60%) and having low self complementarily. Polymerase chain reactions (PCR) used for validating the amplification primers were performed in a total volume of 25 μL containing 50 ng/μL gDNA, GoTaq® Green master mix, 2×, 10 μM primer and nuclease-free water (ddH2O). Thermal cycling conditions were as follows: an initial denaturation step of 95°C for 2 min, then 35 cycles at 95°C for 1 min, 55–60°C for 1 min, 72°C for 1 min, 72°C for 5 min final extension and at 4°C final holding temp. PCR products were loaded directly into 1% agarose gels or non-denaturing TBE polyacrylamide gels. The amplification primers were also assessed using qPCR melting curve analysis.
Table 1. Primers designed for qPCR based on bovine reference genome
2.4. Copy number variation analysis of KLF6, CHKB, GPC1 and CHRM3 genes
Primers designed for copy number variation is listed in Table . The relative copy number variations of the bovine KLF6, CHKB, GPC1 and CHRM3 genes were examined using quantitative real-time polymerase chain reaction (qPCR). Bovine basic transcription factor 3 (BTF3) was used as a normal (two copy or diploid) internal reference gene for genomic qPCR because in the Database of Genomic Variants, neither CNV nor segmental duplication has been described (Bickhart et al., Citation2012). The qPCR reaction was carried out using SYBR® Premix Ex Taq TM II (Tli RNase H Plus) in 25 μL total reaction mixture containing 50 ng of gDNA, 12.5 μL SYBR Premix Ex Taq II (2×) and 10 pmol of primers (TAKARA BIO INC.). The thermal cycling profile of qPCR experiments was: one cycle of 95°C for 1 min followed by 39 cycles at 95°C for 10 s, annealing at 60°C for 30 s, extension at 68°C for 10 s. During qPCR thermal cycles, primers were also examined using melting curve (dissociation) steps and no template control (a minus sample control) included. Experiments were repeated three times, and data used for statistical analyses was obtained using the average mean values and standard deviation.
2.5. Statistical analysis
Copy number was calculated based on average threshold cycle (ΔCt) methods (Schmittgen & Livak, Citation2008), related to reference gene BTF3 (Liu et al., Citation2010). The final result of the copy number was defined using the formula 2 × 2–ΔΔCt (Bae et al., Citation2010), where ΔCt was the difference in threshold cycles for each sample normalized to reference gene (Bae et al., Citation2010). The normal value of DNA copy number in tested animals as diploid and all measured target DNA copy number values were adjusted relative to the two copies (Yim et al., Citation2011). The populations genetics of different CNV types of KLF6, CHKB, GPC1 and CHRM3 genes were grouped into gain (>0.5), loss (<–0.5) and normal (<|±0.5|) of the CNVs based on log2 ratio (log22-△△Ct) relative to control sample previously described elsewhere (Liu et al., Citation2016; Shi et al., Citation2015; Xu et al., Citation2013). The pairwise comparison and scatter plots were determined using GraphPad (PRISM5) statistical software using the value obtained from the log2 ratio (log22-△△Ct).
3. Results
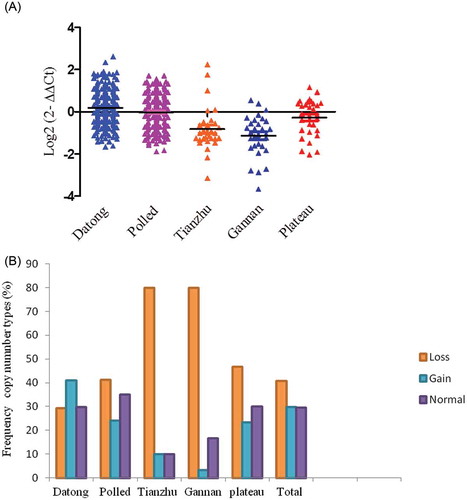
Yak breeding is an economically important sector and supports millions of people for milk and beef; the breeding of yak has been supported intensively by farm animal’s genetic resources, an institute of Chinese academy of agricultural science. Recently, much attention has been paid to the investigation of genomic markers related to population genetics due to a vital role in the livestock selection. As shown in Figure , the CNV of CHKB gene in the Tianzhu, Gannan and plateau yak individuals had greater copy number loss while Datong and polled yak individuals had greater copy number gains. Significant differences were found in the pairwise comparison between Polled versus Tianzhu (P = 0.0001), Gannan (P = 0.0001); Datong versus Tianzhu, Gannan, and Gannan versus Plateau yak (P = 0.0001, P < 0.01). No significant differences were detected between Datong versus Polled (P = 0.35), Plateau (P = 0.013); Polled versus Plateau (P = 0.08) and Tianzhu versus Gannan (P = 0.22), Plateau (P = 0.039) yak in terms of the CNVs of the CHKB gene (Table ). Using both GraphPad (PRISM) and log2 (ratio) analysis, the CNV of CHKB gene differentiates the yak breeds in two distinct groups based on provincial locations or sites, which include Datong, polled and plateau yak in Qinghai province and Tianzhu and Gannan Gansu province. For all breeds, an overall frequency of CHKB copy number loss (copy number 0 or 1), normal level (copy number 2), and gain (more than two copies) was 40.8%, 29.5%, and 29.7%, respectively (shown in Figure ). Approximately 80% of Tianzhu yaks, 46.7% of plateau yaks and 80% of Gannan yaks breeds exhibited loss copy number genotypes, while about 41% of Datong and 23% of polled yak demonstrated copy number gain genotypes (Figure , Table ). The normal copy number genotype was found at higher frequency in the polled (35%) and Datong (29.5%) yak populations.
Table 2. Distribution of the CHKB gene CNV types among five yak breeds sampled from across China
Table 3. Pairwise comparison of CNV of the CHKB gene in five Chinese domestic yak breeds
Figure 2. Distributions of the CNV of CHKB gene in five yak breeds. (A) Log2 ratio the grouped scatter plot distribution of the CNVs in five yak breeds which were constructed using GraphPad (PRISM) (n = 475). (B) The frequency of individuals with different relative copy numbers (0 and 1 = loss, 2 = normal and >2 = gain copy number types). Copy numbers were rounded to the nearest integer number.
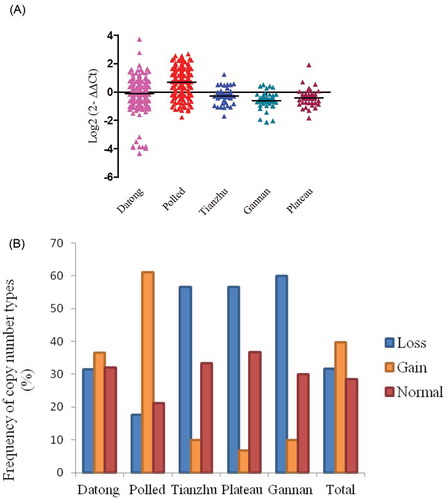
As presented in Figure , the CNV of the KLF6 gene showed greater copy number loss in Tianzhu, Gannan and plateau yak individuals, whilst greater copy number gain was observed in Datong and polled yak individuals. There were highly significant differences in the pairwise comparison between Polled versus Tianzhu, Gannan, Datong, and Plateau yak (P = 0.0001, Table ), however, no significant difference was observed between Datong versus Plateau (P = 0.10), Tianzhu (P = 0.35), Tianzhu versus Gannan (P = 0.085), plateau (P = 0.57) and Gannan versus plateau (P = 0.20) yak breeds (Table ). However, the frequency of copy number loss in KLF6 stood at 31.7% (0 or 1 copy number), normal 28.5% (2 copy numbers) and gain 39.8% (greater two copy numbers) as presented in Figure and Table . Among the populations, 56.7% of Tianzhu, 56.7% of plateau and 60% of Gannan yak breeds contained only loss copy number genotypes. However, 36.5% of Datong and 62.1% of Polled yak contained gain copy number genotypes (Figure and Table ).
Table 4. Distribution of the KLF6 gene CNV types among five yak breeds sampled from across China
Table 5. Pair-wise comparison of CNV of the KLF6 gene in five Chinese domestic yak breeds
Figure 3. Distributions of the CNV of KLF6 gene in five yak breeds. (A) Log2 ratio the grouped scatter plot distribution of the CNVs in five yak breeds which were constructed using GraphPad (PRISM) (n = 477). (B) The frequency of individuals with different relative copy numbers (0 and 1 = loss, 2 = normal and >2 = gain copy number types). Copy numbers were rounded to the nearest integer number.
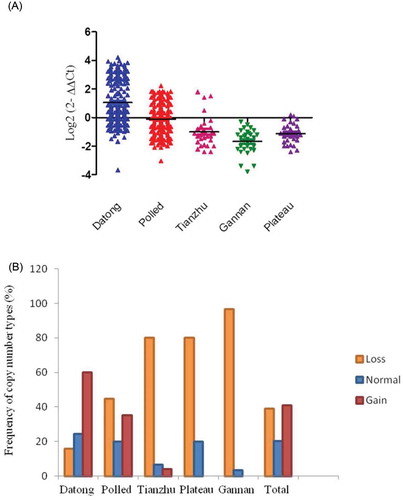
As shown in Figure , the GPC1 gene CNV in the Tianzhu, Gannan and Plateau yak individuals exhibited greater copy number loss while polled yak and Datong yak individuals exhibited copy number gain. The pairwise comparison between Datong versus polled, Tianzhu, Gannan, Plateau yak, and Polled versus Tianzhu, Gannan, Plateau yak breeds was highly significant with a P-value of 0.0001 (P < 0.01, Table ) and a non significant difference was observed in Tianzhu versus Gannan yak breeds (P = 0.14), plateau versus Tianzhu (P = 0.58), and Gannan versus plateau (P = 0.11) yak breeds. Across all breeds, there was an overall frequency of GPC1 copy number (Figure and Table ) loss (copy number 0 or 1), normal level (copy number 2), and gain (more than two copies) of 39%, 20.1%, and 40.9%, respectively. Among them, 80% of Tianzhu, 80% of Plateau, 96.7% of Gannan and 44.8% polled yak breeds contained only loss copy number genotype while 59.9% of Datong yak contained only gain copy numbers genotypes (Figure and Table ).
Table 6. Distribution of the GPC1 gene CNV types among five yak breeds sampled from across China
Table 7. Pair-wise comparison of CNV of the GPC1 gene in five Chinese domestic yak breeds
Figure 4. Distributions of the CNV of GPC1 gene in five yak breeds. (A) Log2 ratio the grouped scatter plot distribution of the CNVs in five yak breeds which were constructed using GraphPad (PRISM) (n = 477). (B) The frequency of individuals with different relative copy numbers (0 and 1 = loss, 2 = normal and >2 = gain copy number types). Copy numbers were determined by real-time PCR assays and rounded to the nearest integer number.
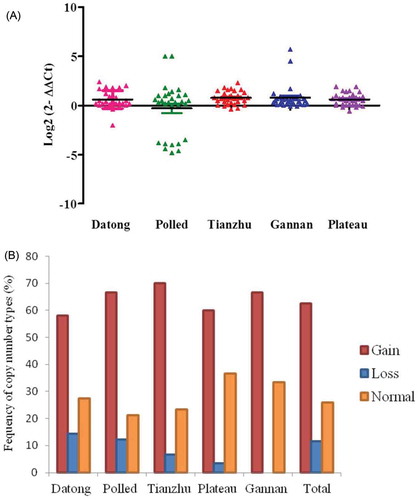
As shown in Figure , the CNV of the CHRM3 gene in polled, Datong, Tianzhu, Gannan and plateau yak individuals exhibited more copy number gains. No significant differences were observed between the five yak breeds when compared with individual populations (Table ). Among the populations, 70% of Tianzhu, 66.7% of Gannan, 58.1% of Datong, 66.7% of polled and 60% of plateau yak breeds contained only gain copy numbers genotypes (Figure and Table ). As shown in Figure , the five types of CHRM3 CNVs showed similar distributions.
Table 8. Distribution of the CHRM3 gene CNV types among five yak breeds sampled from across China
Table 9. Pair-wise comparison of CNV of the CHRM3 gene in five Chinese domestic yak breeds
Figure 5. Distributions of the CNV of CHRM3 gene in five yak breeds. (A) Log2 ratio the grouped scatter plot distribution of the CNVs in five yak breeds which were constructed using GraphPad (PRISM) (n = 30). (B) The frequency of individuals with different relative copy numbers (0 and 1 = loss, 2 = normal and >2 = gain copy number types). Copy numbers were rounded to the nearest integer number.
4. Discussion
Copy number variations contribute significantly to the genetic makeup of a species and have become a valuable resource for investigating population genetics of animal and plant species. CNVs surveying has been completed for most of the domestic animals including yak breeds (Zhang et al., Citation2016). The considerable genetic differences found between the Black Angus and Holstein cattle in CNVs have been investigated using read depth platforms (Stothard et al., Citation2011). Similarly, Fadista, Thomsen, Holm, and Bendixen (Citation2010) reported that a high diversity of CNVs was detected in beef and dairy cattle breeds. Furthermore, Liu et al. (Citation2010) and Hou et al. (Citation2011) confirmed that differences in the CNV frequencies across diverse breeds of cattle; suggesting that some cattle CNVs are likely to have arisen independently within breeds and could contribute to observed breeding differences. Similarly, Bickhart et al. (Citation2012) suggested that CNV differences were found between taurine and indicine cattle breeds reflected their breeding history. Hou et al. (Citation2011) also found that differences between CNV variations, in some cattle breeds, is possibly due to genetic bottlenecks, caused through both selection and genetic drift. Interestingly, a comparative analysis revealed potential contributions of CNVs to the process of yak domestication and significantly different copy number were present between domestic and wild yak animals (Zhang et al., Citation2016). However, most of the studies focusing on the detection CNVRs in domesticated and wild yak population are based on whole genomic sequencing, while, CNVs-based population genetics of yak breeds diversity have not been adequately investigated.
As shown in Figures , , and , the CNVs of the CHKB, KLF6 and GPC1 genes had greater frequency copy number losses in the Tianzhu, Gannan and plateau yak populations while Datong and polled yak showed a higher frequency of copy gains. However, the CHRM3 CNV showed a significantly higher frequency of gain in the five yak populations. These results concur with a previous finding in which Upadhyay et al. (Citation2017) observed that a higher number of genic deletions, as well as higher cumulative length of the genome under deletion in the British and Irish cattle breeds, compared to breeds from across European cattle. The results obtained in Upadhyay’s study could be explained through changes in the past effective population size, gene flow, selection process (purifying selection) and genetic drift, all which may contribute to shaping CNV diversity that existed between the different European cattle populations (Upadhyay et al., Citation2017). The present results have suggested that the CNVs in the CHKB, KLF6, GPC1 and CHRM3 genes are associated with yak breed formation. Our results showed that Tianzhu yaks had high genetic variability within a population, due to intensive selection program for white hair individuals (Zhang et al., Citation2008); for the reason that the white hair is easily dyed into different colors and highly valued in markets, the nomadic pastoralists migrated from Qinghai province and started to select and breeding pure white herds about 120 years ago in Tianzhu County (Wiener et al., Citation2003). In addition, Yue, Liang, Liang, and Li (Citation2016) observed that the Tianzhu white yak possessed relatively high genetic diversity resulting from hybridization with Bos Taurus. Our results correlated positively to previous findings in which the genetically isolated small populations have been found to accumulate an abundance of deletion type CNVs (Upadhyay et al., Citation2017). Similarly, within Chinese cattle breeds, most individuals have higher frequencies of copy number losses in the MYH3, LEPR and MAPK10 genes (Liu et al., Citation2016; Shi et al., Citation2015; Xu et al., Citation2013). Interestingly, an earlier study of CNVs in the Bos taurus UL16-binding protein (ULBP) gene found that the highest copy number gain and loss were detected in Limousin and Angus cattle breeds, respectively, due to a combination of genetic bottlenecks in some breeds, selection, and genetic drift (Liu et al., Citation2010). Population differentiation in this study was discovered using log2 ratio resultant to copy number gains, normal and losses (Fadista et al., Citation2010; Liu et al., Citation2016; Olshen, Venkatraman, Lucito, & Wigler, Citation2004). The differences in CNV frequencies in the five yak breeds, may reflect the breed variation between individual populations (Lehnert et al., Citation2007; Yang et al., Citation2017) as differences in CNV frequency amongst cattle breeds is related to breed formation and adaptation (Liu et al., Citation2010). The data in this study was analyzed using log2 ratio to differentiate the genetic diversity between yak breeds, which appears to be more sensitive than the detection of homozygous deletions (Liu et al., Citation2016; Olshen et al., Citation2004). According to Fadista et al. (Citation2010) findings, the biological and technical detection methods of CNVs are biased towards deletions, and more deletions than duplications have been produced by mechanisms of non-allelic homologous recombination (NAHR) (Turner et al., Citation2008). The results obtained in this present research, is in agreement with the findings by Fadista et al. (Citation2010), in which log2 ratio exhibited more powerful detection of loss than gain copy number types. Lee et al. (Citation2013) revealed that significant copy number variations in the OR gene were found in Korean native cattle, Black Angus and Holstein, indicating that there is selection pressure maintaining the integrity of OR genes. In addition, the CNVRs of the OR gene is different in Angus, Red Angus, and Holstein cattle and CNVRs of the sterol carrier protein 2 (SCP2) gene is different in Gelbvieh and Red Angus (Liu et al., Citation2010). Perry et al. (2007) suggested that natural selection has contributed in shaping AMY1 copy number variation in both Japanese and Yakut populations. In human, high copies of the AMY1 number were reported as compared to other primates, which have been attributed to the dietary shift that occurred early in the hominid evolutionary history (Perry et al., 2007). The human UDP glucuronosyltransferase family two member B17 (UGT2B17) gene varies in copy number from zero to two per individual from Africa, Europe, and East Asia people (Xue et al., Citation2008). This variation is a result of positive selection and evolutionary change over the last centuries. The deletion in the UGT2B17 CNVs is relatively rare in African and European populations and common in East Asian populations (Xue et al., Citation2008). The CNVs of the UGT2B17 and AMY1 genes have been hypothesised to have a crucial role in population differentiation (Fernández & Wiley, Citation2017; Perry et al., 2007; Xue et al., Citation2008). Xu et al. (Citation2016) indicated that CNV deletions showed large differences in frequency across diverse groups of cattle. Intriguingly, Olson (Citation1999) mentioned that gene loss was a major pattern of molecular evolution and is a possible evolutionary pressure that may explain the geographic pattern (Ko et al., Citation2012), hence, this combination may play a prominent role in molecular evolution and function (Gamazon & Stranger, Citation2015). Correspondingly, Zarrei, MacDonald, Merico, and Scherer (Citation2015) found that CNVs caused by deletions and duplications in the genome were the predominant causes of the variability among populations, with duplications and deletions occurring as results of positive and negative selective pressure in the individual populations, respectively.
The distribution of CNVs for the CHKB, KLF6 and GPC1 genes showed a similar pattern across all five yak breeds. Copy number distributions of the CHKB and GPC1 genes were not in agreement to previous analysis of Jincheng, Zhihua, Sujun, and Yuping (Citation2006) using a analysis of microsatellite markers, RAPD, AFLP, chromosomes and blood protein polymorphisms methods showing that Datong and the Tianzhu white yaks clustered together. Copy numbers of the KLF6 and CHRM3 genes, on the other hand, were consistent with previous findings of Jincheng et al. (Citation2006). A high degree of genetic diversity in the Tibetan yak populations in relation to mtDNA COIII gene has been reported (Song et al., Citation2015). Moreover, the results of the distribution of copy number of the CHKB and GPC1 genes strongly supported the hypothesis proposed by Bao (Citation2016) that the Datong and polled clustere together while Gannan yak clustere into a separate clade, consistent with the known yak breed history. The current results support the work of Liang et al. (Citation2016) who showed that the pairwise distances between Datong and polled yaks were not larger than within population and there was no significant differentiation between the two breeds when the phylogenetic tree was constructed using whole genome-wide association study. However, SNPs studies have confirmed that most polled yak individuals were heterozygous compared to Datong individuals (Liu et al., Citation2013). A recent study has demonstrated the detection of five different haplotypes within the Datong and polled yak populations, and the haplotype frequency that was differently distributed between the polled and Datong yaks (Liu et al., Citation2013). However, the level of nucleotide diversity is higher in the polled yaks than in the Datong yaks (Liu et al., Citation2013). Hence, Liang et al. (Citation2016) suggested that expression variations rather than structural variations in the encoded protein probably contributed to the polled phenotype.
However, unlike previous studies using mitochondrial proteomics (Bao, Citation2016) and other markers (Jincheng et al., Citation2006) the breeds from the same region or ecotype did not cluster together, for instance, Datong, polled and plateau yak in the CNVs of the KLF6 and CHRM3 genes (Figures and ). This indicated that, there was relatively low level of divergence in copy number of the KLF6 and CHRM3 genes within the different yak breeds. On the other hand, Datong, plateau, Tianzhu, and Gannan yak populations were all clustered together, whereas the polled population clustered alone in CNVs of the KLF6. The plateau, Tianzhu, and Gannan yak populations were clustered together while Datong and polled yak populations were clustered with other groups in CNVs of GPC1. In relation to CHRM3 gene, we detected no variation. These results imply that the classifications of yak populations are mainly based on geographic distribution, ecosystem, conditions, the cultivation and differentiation of the breeds (Jincheng et al., Citation2006). Similarly, clustering based on CNVs of the aldehyde oxidase 3 (APOL3) and aldehyde oxidase 1 (AOX1) genes in cattle has been done in line to the evolutionary history of the breeds (Bickhart et al., Citation2012). Similarly, based on nuclear microsatellite markers, Zhang et al. (Citation2008) revealed that 94.4% of the genetic variation was observed within yak breeds, while only 5.6% of the genetic variation existed between breeds. In addition, studies performed by Wang et al. (Citation2010), support our findings, in which 93.91% total variance within populations, 5.46% among populations, and 0.64% among regions in domestic yaks were observed by use of mitochondrial DNA analysis (Wang et al., Citation2010). The results obtained by Wang et al. (Citation2010), strongly support the theory that variations due to genetic diversity, genetic distance, and cluster analyses among the Tibetian yak populations are not directly correlated to geographical distribution. This supports the findings of several studies which have shown that the effect of geographical environment on genetic relationships between yak populations has gradually decreased over time (Song et al., Citation2015; Wang et al., Citation2008; Zhang et al., Citation2008). Thus, low sample size per breed does not follow the same pattern of demographic events in the history of yak domestication, as previously observed in cattle population (Upadhyay et al., Citation2017).
It’s perhaps a surprising observation that the separation and clustering of the cattle using CNVs is not superior to those based on SNPs and suggests that CNV genotyping still has room for improvement (Bickhart et al., Citation2016). In addition, Upadhyay et al. (Citation2017) speculated that de-novo CNVs, CNV hot-spot regions in the genome and false CNV calls due to variation in the genotyping intensities also affect inference of population stratification. Indeed, Xu et al. (Citation2016) suggested the CNVs have been used to separate cattle individuals into distinct groups, however, the clustering resolution within groups based on CNVs was not better than those based on SNPs. For instance, CNVs cannot distinguish the Holstein breed from the Angus breed in European taurine cattle and certain degrees of mixing within indicine individuals from CNV-based clustering results (Xu et al., Citation2016). Therefore, due to the accuracy of genotyping of SNPs, CNV based population stratification can be only detected among the large population groups at the continent level (Jakobsson et al., Citation2008; Redon et al., Citation2006; Upadhyay et al., Citation2017; Xu et al., Citation2016). Similarly, Liu et al. (Citation2010) and Hou et al. (Citation2011) described that more CNV were detected in indicine (17.63) and composite (15.75) than in taurine breeds (10.73), while within the taurine breeds, more CNVs were found in African breeds (11.60) than in European breeds (10.63), suggesting that differences CNVs among cattle breeds may be linked to the reference sample of Hereford cow of European origin, breed divergence, and history. It was intriguing to note that gene ancestries, structures, functions, and genomic distributions, gene duplication, positive selection, and conversion have shaped CNVs (Liu et al., Citation2010).
In addition, the within-population variability of Tianzhu, Gannan, and plateau yak breeds compared to Datong and polled yak breeds in CHKB, KLF6 and GPC1 copy number may be attributed to the high admixture pattern of their genomes (Qi, Jianlin, Wang, Rege, & Hanotte, Citation2009). High admixture of yak breeds in the Qinghai-Tibetan Plateau, Mongolian and Russian regions, however, a low admixture of yak breeds in the Himalayan and Pamir Plateau region have been reported (Qi et al., Citation2009). Such difference of copy number between different populations has also been reported in other species (Bickhart et al., Citation2012; Liu et al., Citation2010; Xu et al., Citation2016). The difference in CNV genes between the yak breeds may reflect gene flow, selection, and genetic drift. The frequencies of cattle introgression ranging from 1.5% Tianzhu white yak to 10.6% Gannan black yak has been shown using mt-DNA (Xuebin, Jianlin, Rege, & Hanotte, Citation2002). Zhang, Xu, et al. (Citation2014) indicated that genetic variations coat colors of yak are probably due to natural (evolution and domestication) and artificial (interspecific hybridization, i.e. natural and artificial) selection. This may be the cause of some of the observed genetic variability among yak individuals. Recent studies have reported several examples of gene flow and introgression from cattle to yaks (Qi et al., Citation2009), the introgressed regions are enriched in genes involved in nervous system development and function, and particularly in glutamate metabolism and neurotransmission and may contribute to the improvement of yak management and breeding (Medugorac et al., Citation2017). In contrast, Nguyen et al. (Citation2005) suggested that there was no significant differentiation between the Datong and Gannan yak populations using bovine microsatellite markers. As outlined above, the CNVs of yak populations with high diversity within individuals were described previously (Qiu et al., Citation1986; Zhang, Citation1989). The northern Tibet and eastern Qinghai Tibetan Plateau areas were areas of domestication and the origin of the yak breeds (Jincheng et al., Citation2006). These findings are explained by Hanotte et al. (Citation2002) where higher diversity indices result from the retention of greater numbers of alleles from the original domestication event and the least loss of genetic diversity closest to the center of origin of domestication during the migration of domestic animals. Similarly, genome CNV evolution and adaption may be a potent evolutionary force for more loss events (Zhang, Jia, et al., Citation2014). These results indicated that in the yak population, there are several important genes related to clear phenotypic changes and breed differences have been modified by CNVs and gene modulations. The high degree of phenotypic and genomic differences between breeds is partly due to a long domestication history and adaptation to different local and climatic conditions throughout China (Qi et al., Citation2005; Zhang et al., Citation2008). CNVs are significant effects on genomic sequences due to changing gene structure and dosage, alternating gene regulation and exposing recessive alleles (Henrichsen, Chaignat, & Reymond, Citation2009; Zhang, Gu, Hurles, & Lupski, Citation2009). Interestingly Liu et al. (Citation2010) reported that the difference of CNV among the cattle breeds may coincide with multiple independent domestications of cattle in the Fertile Crescent, Southwest Asia, and Africa (Caramelli, Citation2006; Troy et al., Citation2001). Wiener et al. (Citation2003) observed that there is frequent exchange of yak breeds and migration of herdsmen in yak habitat areas of China; hence, distribution centers of certain Chinese yak breeds have been changed corresponding to climate change over the past 50 years (Wu, Citation2016). Our study suggests the need for further investigation using larger yak population sizes to further validate and improve CNV genotyping approaches.
The present results show that the copy number gain events may be due to extra genetic material (artificial insemination, hybridization, and introgression) obtained during breed formation in Datong and Polled yak, since Datong yaks are produced by mating Huanhu yak cows with wild yaks (male) for meat production and Polled yak are produced from breeding Polled yak cows with horned yak bulls (Liu et al., Citation2013; Wiener et al., Citation2003); the other three yak breeds belong to the indigenous domestic yaks of China. In agreement with this results, CNV differences among cattle breeds due to altered metabolic and immune requirements, microbial fermentation in the rumen, the herd environment, and the reproductive strategy of cattle through human selection (Liu et al., Citation2010). In agreement with our results, Stothard et al. (Citation2011) found that strong selection in Holstein cattle breeds in the past 50 years has led to high copy number gains and shaping CNVs. The current results suggested that a very low level of genetic diversity is present in the Chinese domestic yak breeds. Similar results had been reported in the Chinese cattle (Shi et al., Citation2015), for CNVs of LEPR gene. Interestingly, the CNV distribution of the KLF6 gene in the Datong yak breed, the CNV distribution of the CHKB and GPC1 genes in the polled and Datong yak breeds, and the CNV distribution of the CHRM3 gene in the Datong, polled, Tianzhu, plateau and Gannan yak breeds seems to be more centralized than that for other breeds. Beckmann, Estivill, and Antonarakis (Citation2007) reported that continued characterization of genetic CNVs, particularly between breeds, could be an important step towards deciphering the effects of trait variations.
5. Conclusions
The development of new techniques at molecular levels have help in resolving the population genetics dynamics and more among the closely related organisms (Dufresne, Stift, Vergilino, & Mable, Citation2014). CNVs is a more recent technique has been successfully applied in various studies for instance, in the analysing their effect on gene expression and their association with disease susceptibility and other complex human traits (Gamazon, Nicolae, & Co, Citation2011). In this study, we applied the CNVs in determining the variation among the yaks in China. The results showed that the CNVs of the CHKB, KLF6, and GPC1 genes presented more copy number losses in Tianzhu, Gannan and Plateau yak populations while Datong and polled yak more copy number gains. However, the CHRM3 CNV was more copy number gains in five yak populations. Therefore, these results support the hypothesis that log2 ratio is more powerful detection loss than gain copy number types. It is interesting, unlike previous studies, the breeds from the same provincial did not cluster together using CNVs of different genes, due to the different diversity of individuals and the technology used for detection. In the present results shown that the copy number gain events might be due to artificial insemination, hybridization, and introgression of breeds during breed formation. Taken together, these observations suggest that the CNVs of the CHKB, KLF6, GPC1 and CHRM3 genes could exert a significant effect on phenotypic differences in yak populations, highlighting their diversity in terms of CNVs. This study was based on real-time polymerase chain reaction (qPCR) using five different domestic yaks; therefore, it is difficult to ascertain the CNVs of genes in other studies due to the different populations, sizes, platforms, and algorithms applied for surveying.
Ethics statement
The study was conducted between September 2016 and December 2017 at Key Laboratory of Yak Breeding Engineering of Gansu Province, Lanzhou Institute of Husbandry and Pharmaceutical Sciences, the data obtained from yak breeding cooperatives of Gansu and Qinghai Province, China. All blood sample collection and body measurements were strict accordance of the guide for the Care and Use of Laboratory Animals, Lanzhou Institute of Husbandry Animal and Pharmaceutical Sciences, China. Also, all animals were slaughtered under anesthesia, and all necessary efforts were made to minimize risk of suffering. Thus, we agree to perform the research on the yak and the legal certificate number was SCXK (Gan) 2014-0002.
Competing interests
The authors declare no competing interests.
Author contributions
Habtamu Abera, data analysis, reviewed different journals and books in the line of this manuscript and Professor Ping Yan and Dr. Bao Pengjia, contributed to its supervisory and guiding. Dr. Wu Xiaoyun involved in drafting the manuscript and laboratory works. Habtamu made all writing and sketch of the manuscript.
Cover image
Source: Habtamu Abera Goshu (2017), Key Laboratory for Yak Genetics, Breeding & Reproduction Engineering of Gansu Province, Chinese Academy of Agricultural Science.

Acknowledgements
We thank Mr. Yong Feng Zhang for guidance and technical assistance during the laboratory work and Dr. Hong Bo Wang, Ms. Li Xiaoxiao and Mr. Peng Tang for blood sample collection.
Additional information
Notes on contributors

Habtamu Abera Goshu
Ping Yan is a specialist in Genetics, Breeding & Reproduction. She has dedicated herself to the topics related to yak genetic resource and breeding. Her current research focuses primarily on evaluation and application of new developments and new technologies in sustainable yak breeding and reproduction, and marker-assisted selection (MAS) to increase disease resistance, productivity and product quality in economically important animals by adding information of DNA markers to phenotypes and genealogies for selection decisions. Currently, she is the deputy director of Lanzhou Institute of Husbandry and Pharmaceutical Sciences and director of Key Laboratory for Yak Genetics, Breeding & Reproduction Engineering of Gansu Province, Chinese Academy of Agricultural Science. She was successfully bred a new “strain” of Datong yak, which filled the blank in the history of the world 2019s domesticated animal breeding, therefore she has independent intellectual property right.
References
- Almal, S. H., & Padh, H. (2012). Implications of gene copy-number variation in health and diseases. Journal of Human Genetic, 57, 6–13. doi:10.1038/jhg.2011.108
- Bae, J. S., Cheong, H. S., Kim, L. H., NamGung, S., Park, T. J., Chun, J.-Y., … Shin, H. D. (2010). Identification of copy number variations and common deletion polymorphisms in cattle. BMC Genomics, 11, 232. doi:10.1186/1471-2164-11-232
- Bao, P. (2016). Integrative analysis of muscle mitochondria proteomic in yak hypoxic adaptation (doctoral dissertation abstract). Chinese Academic of Agricultural Science, Lanzhou, China.
- Beckmann, J. S., Estivill, X., & Antonarakis, S. E. (2007). Copy number variants and genetic traits: Closer to the resolution of phenotypic to genotypic variability. Nature Reviews Genetics, 8, 639–646.
- Bickhart, D. M., Hou, Y., Schroeder, S. G., Alkan, C., Cardone, M. F., Matukumalli, L. K., … Liu, G. E. (2012). Copy number variation of individual cattle genomes using next-generation sequencing. Genome Research, 22(4), 778–790. doi:10.1101/gr.133967.111
- Bickhart, D. M., Xu, L., Hutchison, J. L., Cole, J. B., Null, D. J., Schroeder, S. G., … Liu, G. E. (2016). Diversity and population-genetic properties of copy number variations and multicopy genes in cattle. DNA Research: an International Journal for Rapid Publication of Reports on Genes and Genomes, 23(3), 253–262. doi:10.1093/dnares/dsw013
- Caramelli, D. (2006). The origins of domesticated cattle. Human Evolution, 21, 107–122.
- Chen, C., Qiao, R., Wei, R., Guo, Y., Ai, H., Ma, J., … Huang, L. (2012). A comprehensive survey of copy number variation in 18 diverse pig populations and identification of candidate copy number variable genes associated with complex traits. BMC Genomics, 13, 733. doi:10.1186/1471-2164-13-733
- Conrad, D. F., Pinto, D., Redon, R., Feuk, L., Gokcumen, O., Zhang, Y., … Hurles, M. E. (2010). Origins and functional impact of copy number variation in the human genome. Nature, 464(7289), 704–712. doi:10.1038/nature08516
- Cook, E. H., Jr., & Scherer, S. W. (2008). Copy-number variations associated with neuropsychiatric conditions. Nature, 455(7215), 919–923.
- Dufresne, F., Stift, M., Vergilino, R., & Mable, B. K. (2014). Recent progress and challenges in population genetics of polyploidy organisms: Anoverview of current status –Of-the-art molecular and statistical tools. Molecular Ecology, 23(1), 40–69. doi:10.1111/mec.12581
- Fadista, J., Thomsen, B., Holm, L.-E., & Bendixen, C. (2010). Copy number variation in the bovine genome. BMC Genomics, 11, 284. doi:10.1186/1471-2164-11-284
- Fernández, C. I., & Wiley, A. S. (2017). Rethinking the starch digestion hypothesis for AMY1 copy number variation in humans. American Journal of Physical Anthropology, 163(4), 645–657. doi:10.1002/ajpa.23237
- Freeman, J. L., Perry, G. H., Feuk, L., Redon, R., McCarroll, S. A., Altshuler, D. M., … Lee, C. (2006). Copy number variation: New insights in genome diversity. Genome Research, 16(8), 949–961. doi:10.1101/gr.3677206
- Gamazon, E. R., Nicolae, D. L., & Co, N. J. (2011). A study of CNVs as traits- assocated polymorphisum and as expression quantitative traits loci. Plos Genetics, 7(2), e1001292. doi:10.1371/journal.pgen.1001292
- Gamazon, E. R., & Stranger, B. E. (2015). The impact of human copy number variation on gene expression. Briefings in Functional Genomics, 14(5), 352–357. doi:10.1093/bfgp/elv017
- Girirajan, S., Dennis, M. Y., Baker, C., Malig, M., Coe, B. P., Campbell, C. D., … Eichler, E. E. (2013). Refinement and discovery of new hotspots of copy-number variation associated with autism spectrum disorder. American Journal of Human Genetics, 92(2), 221–237. doi:10.1016/j.ajhg.2012.12.016
- Hanotte, O., Bradley, D. G., Ochieng, J. W., Verjee, Y., Hill, E. W., & Rege, J. E. O. (2002). African pastoralism: Genetic imprints of origins and migrations. Science, 296, 336−339. doi:10.1126/science.1069878
- Henrichsen, C. N., Chaignat, E., & Reymond, A. (2009). Copy number variants, diseases, and gene expression. Journal of Human Molecular Genetics, 18(R1), R1–8. doi:10.1093/hmg/ddp011
- Hou, Y., Liu, G. E., Bickhart, D. M., Cardone, M. F., Wang, K., Kim, E., … Van Tassell, C. P. (2011). Genomic characteristics of cattle copy number variations. BMC Genomics, 12, 127. doi:10.1186/1471-2164-12-127
- Iafrate, A. J., Feuk, L., Rivera, M. N., Listewnik, M. L., Donahoe, P. K., Qi, Y., … Lee, C. (2004). Detection of large-scale variation in the human genome. Nature Genetics, 36(9), 949–951. doi:10.1038/ng1416
- Iskow, R. C., Gokcumen, O., & Lee, C. (2012). Exploring the role of copy number variants in human adaptation. Trends in Genetics: TIG, 28(6), 245–257. doi:10.1016/j.tig.2012.03.002
- Jakobsson, M., Scholz, S. W., Scheet, P., Gibbs, J. R., VanLiere, J. M., Fung, H. C., & Singleton, A. B. (2008). Genotype, haplotype and copy-number variation in worldwide human populations. Nature, 451(7181), 998–1003. doi:10.1038/nature06742
- Jincheng, Z., Zhihua, C., Sujun, Z., & Yuping, X. (2006). Classification of ecological types of the Chinese yak. Acta Ecologica Sinica, 26, 2068–2072.
- Kijas, J. W., Townley, D., Dalrymple, B. P., Heaton, M. P., Maddox, J. F., & McGrath, A.; for the International Sheep Genomics Consortium. (2009). A genome wide survey of SNP variation reveals the genetic structure of sheep breeds. PLoS ONE, 4(3), e4668.
- Ko, D. C., Gamazon, E. R., Shukla, K. P., Pfuetzner, R. A., Whittington, D., Holden, T. D., … Miller, S. I. (2012). Functional genetic screen of human diversity reveals that a methionine salvage enzyme regulates inflammatory cell death. Proceedings of the National Academy of Sciences of the United States of America, 109(35), E2343–E2352. doi:10.1073/pnas.1206701109
- Lee, K., Nguyen, D. T., Choi, M., Cha, S.-Y., Kim, J.-H., Dadi, H., … Park, C. (2013). Analysis of cattle olfactory subgenome: The first detail study on the characteristics of the complete olfactory receptor repertoire of a ruminant. BMC Genomics, 14, 596. doi:10.1186/1471-2164-14-596
- Lehnert, S. A., Reverter, A., Byrne, K. A., Wang, Y., Nattrass, G. S., Hudson, N. J., & Greenwood, P. L. (2007). Gene expression studies of developing bovine longissimus muscle from two different beef cattle breeds. BMC Developmental Biology, 7, 95. doi:10.1186/1471-213X-7-95
- Liang, C., Wang, L., Wu, X., Wang, K., Ding, X., Wang, M., … Yan, P. (2016). Genome-wide association study identifies loci for the polled phenotype in yak. PLoS ONE, 11(7), e0158642. doi:10.1371/journal.pone.0158642
- Liu, G. E., Hou, Y., Zhu, B., Cardone, M. F., Jiang, L., Cellamare, A., … Keele, J. W. (2010). Analysis of copy number variations among diverse cattle breeds. Genome Research, 20(5), 693–703. doi:10.1101/gr.105403.110
- Liu, M., Lia, B., Huanga, Y., Yanga, M., Lana, X., Lei, C., & Chena, H. (2016). Copy number variation of bovine MAPK10 modulates the transcriptional activity and affects growth traits. Livestock Science, 194, 44–50. doi:10.1016/j.livsci.2016.09.014
- Liu, W. B., Liu, J., Liang, C. N., Guo, X., Bao, P. J., Chu, M., & Yan, P. (2013). Associations of single nucleotide polymorphisms in candidate genes with the polled trait in Datong domestic yaks. Animal Genetics, 45(1), 138–141. doi:10.1111/age.12081
- Medugorac, I., Graf, A., Grohs, C., Rothammer, S., Zagdsuren, Y., Gladyr, E., … Capitan, A. (2017). Whole-genome analysis of introgressive hybridization and characterization of the bovine legacy of Mongolian yaks. Nature Genetics, 49(3), 470–475. doi:10.1038/ng.3775
- Nguyen, T. T., Genini, S., Menetrey, F., Malek, M., Vogeli, P., Goe, M. R., & Stranzinger, G. (2005). Application of bovine microsatellite markers for genetic diversity analysis of Swiss yak (Poephagus grunniens). Animal Genetics, 36, 484–489.
- Novembre, J., & Ramachandran, S. (2011). Perspectives on human population structure at the cusp of the sequencing era. Annual Review of Genomics and Human Genetics, 12, 245–274. doi:10.1146/annurev-genome-090810-183123
- Olshen, A. B., Venkatraman, E., Lucito, R., & Wigler, M. (2004). Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics, 5, 557–572.
- Olson, M. V. (1999). When less is more: Gene loss as an engine of evolutionary change. American Journal of Human Genetics, 64(1): 18–23.
- Parsch, J., & Ellegren, H. (2013). The evolutionary causes and consequences of sex-biased gene expression. Nature Reviews Genetics, 14(2), 83–87. doi:10.1038/nrg3376
- Perry, G. H, Dominy, N. J, Claw, K. G, Lee, A. S, Fiegler, H, Redon, R, ... Stone, A. C. (2007). Diet the evoluation of human amylase gene copy number variation. Nature Genetics, 39(10): 1256-1260. doi: 10.1038/ng2123
- Pollex, R. L., & Hegele, R. A. (2007). Copy number variation in the human genome and its implications for cardiovascular disease. Circulation, 115, 3130–3138.
- Qi, X. B., Han, J. L., Lkhagva, B., Chekarova, I., Badamdorj, D., Rege, J. E. O., & Hanotte, O. (2005). Genetic diversity and differentiation of mongolian and russian yak populations. Animal Genetics, 122, 117−126.
- Qi, X. B., Jianlin, H., Wang, G., Rege, J. E. O., & Hanotte, O. (2009). Assessment of cattle genetic introgression into domestic yak populations using mitochondrial and microsatellite DNA markers. Animal Genetics, 41, 242–252.
- Qiu, H., Qin, Z. R., Chen, Y. C., Wang, D. A., Wei, W. Y., Li, K. L., … Cai, L. (1986). Bovine breeds in China. Shanghai: Shanghai Scientific and Technical Publishers.
- Quanwei, Z., Youji, M., Yong, Z., & Xingxu, Z. (2014). Cross-species analysis of copy number variations in domestic yak genome based study on heterologous hybridization using bovine HD genotyping bead chip array. Proceeding of the Fifth international conference on yak, pp.131–138.
- Redon, R., Ishikawa, S., Fitch, K. R., Feuk, L., Perry, G. H., Andrews, T. D., … Hurles, M. E. (2006). Global variation in copy number in the human genome. Nature, 444(7118), 444–454. doi:10.1038/nature05329
- Scherer, S. W., Lee, C., Birney, E., Altshuler, D. M., Eichler, E. E., Carter, N. P., … Feuk, L. (2007). Challenges and standards in integrating surveys of structural variation. Nature Genetics, 39(7 Suppl), S7–15. doi:10.1038/ng2093
- Schmittgen, T. D., & Livak, K. J. (2008). Analyzing real-time PCR data by the comparative CT method. Nature Protocols, 3, 1101–1107.
- Shi, T., Yao, X., Mingjuan, Y., Yongzhen, H., Xianyong, L., Chuzhao, L., … Hong, C. (2015). Copy number variations at LEPR gene locus associated with gene expression and phenotypic traits in Chinese cattle. Animal Science Journal, 87, 336–343. doi:10.1111/asj.12531
- Sjödin, P., & Jakobsson, M. (2012). Population genetic nature of copy number variation. In L. Feuk (Ed.), Genomic structural variants methods in molecular biology (Vol. 2838, pp. 209–223). New York, NY: Springer.
- Song, Q. Q., Chai, Z. X., Xin, J. W., Zhao, S. J., Ji, Q. M., & Zhong, J. C. (2015). Genetic diversity and classification of Tibetan yak populations based on the mtDNA COIII gene. Genetics and Molecular Research, 14(1), 1763–1770. doi:10.4238/2015.March.13.3
- Stankiewicz, P., & Lupski, J. R. (2010). Structural variation in the human genome and its role in disease. Annual Review of Medicine, 61, 437–455. doi:10.1146/annurev-med-100708-204735
- Stothard, P., Choi, J.-W., Basu, U., Sumner-Thomson, J. M., Meng, Y., Liao, X., & Moore, S. S. (2011). Whole genome resequencing of black Angus and Holstein cattle for SNP and CNV discovery. BMC Genomics, 12, 559. doi:10.1186/1471-2164-12-559
- Troy, C. S., Machugh, D. E., Bailey, J. F., Magee, D. A., Loftus, R. T., Cunningham, P., … Bradley, D. G. (2001). Genetic evidence for Near-Eastern origins of European cattle. Nature, 410, 1088–1091.
- Turner, D. J., Miretti, M., Rajan, D., Fiegler, H., Carter, N. P., Blayney, M. L., … Hurles, M. E. (2008). Germline rates of de novo meiotic deletions and duplications causing several genomic disorders. Nature Genetics, 40, 90–95.
- Upadhyay, M., Da Silva, V. H., Megens, H.-J., Visker, M. H. P. W., Ajmone-Marsan, P., Bâlteanu, V. A., … Crooijmans, R. P. M. A. (2017). Distribution and functionality of copy number variation across European cattle populations. Frontiers in Genetics, 8, 108. doi:10.3389/fgene.2017.00108
- Wang, J., Wang, W., Li, R., Li, Y., Tian, G., Goodman, L., ...Wang, J. (2008). The diploid genome sequence of Asian individual. Nature, 456(7218): 60-65.
- Wang, Z., Shen, X., Liu, B., Su, J., Yonezawa, T., Yu, Y., … Liu, J. (2010). Phylogeographical analyses of domestic and wild yaks based on mitochondrial DNA: New data and reappraisal. Journal of Biogeography, 37, 2332–2344. doi:10.1111/j.1365-2699.2010.02379
- Weischenfeldt, J., Symmons, O., Spitz, F., & Korbel, J. O. (2013). Phenotypic impact of genomic structural variation: Insights from and for human disease. Nature Reviews Genetics, 14, 125–138. doi:10.1038/nrg3373
- Wiener, G., Han, J. L., & Long, R. J. (2003). The yak (pp. 189–200). Bangkok, Thailand: Regional Office for Asia and the Pacific of the Food and Agriculture Organization of the United Nations.
- Wu, J. (2016). The distributions of Chinese yak breeds in response to climate change over the past 50 years. Animal Science Journal, 87, 947–958. doi:10.1111/asj.12526
- Xu, L., Hou, Y., Bickhart, D. M., Zhou, Y., Hay, E. H. A., Song, J., … Liu, G. E. (2016). Population-genetic properties of differentiated copy number variations in cattle. Scientific Reports, 6, 23161. doi:10.1038/srep23161
- Xu, Y., Shi, T., Cai, H., Zhou, Y., Lan, X., Zhang, C., & Chen, H. (2013). Associations of MYH3 gene copy number variations with transcriptional expression and growth traits in Chinese cattle. Genetics, 535, 106–111. doi:10.1016/j.gene.2013.11.057
- Xue, Y., Sun, D., Daly, A., Yang, F., Zhou, X., Zhao, M., … Tyler-Smith, C. (2008). Adaptive evolution of UGT2B17 copy number variation. American Journal of Human Genetics, 83(3), 337–346. doi:10.1016/j.ajhg.2008.08.004
- Xuebin, Q., Jianlin, H., Rege, J. E. O., & Hanotte, O. (2002). Cattle mitochondrial DNA introgression in yak (Poephagus or Bos grunniens). Proceedings of the 28th International Conference of Animal Genetics held in Göttingen, Germany, 11-15 August 2002, p. 102 (abstract).
- Yang, M., Lv, J., Zhang, L., Li, M., Zhou, Y., Lan, X., … Chen, H. (2017). Association study and expression analysis of CYP4A11 gene copy number variation in Chinese cattle. Scientific Reports, 7, 46599. doi:10.1038/srep46599
- Yang, Y., Chung, E. K., Wu, Y. L., Savelli, S. L., Nagaraja, H. N., Zhou, B., & Yung, Y. C. (2007). Gene copy-number variation and associated polymorphisms of complement component C4 in human systemic lupus erythematosus (SLE): Low copy number is a risk factor for and high copy number is a protective factor against SLE susceptibility in European Americans. American Journal of Human Genetics, 80, 1037–1054. doi:10.1086/518257
- Yim, S. H., Chung, Y. J., Jin, E. H., Shim, S. C., Kim, J. Y., Kim, Y. S., & Chung, H.-T. (2011). The potential role of VPREB1 gene copy number variation in susceptibility to rheumatoid arthritis. Molecular Immunology, 48, 1338–1343. doi:10.1016/j.molimm.2010.11.009
- Yue, X., Liang, Y., Liang, Y., & Li, F. (2016). Comprehensive investigation of nucleotide diversity in yaks. Animal Genetics, 47(6), 752–755. doi:10.1111/age.12467
- Zarrei, M., MacDonald, J. R., Merico, D., & Scherer, S. W. (2015). A copy number variation map of the human genome. Natural Review Genetics, 16(3), 172–183. doi:10.1038/nrg3871
- Zhang, F., Gu, W., Hurles, M. E., & Lupski, J. R. (2009). Copy number variation in human health, disease, and evolution. Annual Review Genomics of Human, 10, 451–481. doi:10.1146/annurev.genom.9.081307.164217
- Zhang, G. X., Chen, W. S., Xue, M., Wang, Z. G., Chang, H., Han, X., & Wang, D. L. (2008). Analysis of genetic diversity and population structure of Chinese yak breeds (Bos grunniens) using microsatellite markers. Journal of Genetics and Genomics, 35, 233–238.
- Zhang, L., Jia, S., Yang, M., Xu, Y., Li, C., Sun, J., … Chen, H. (2014). Detection of copy number variations and their effects in Chinese bulls. BMC Genomics, 15(1), 480. doi:10.1186/1471-2164-15-480
- Zhang, M. Q., Xu, X., & Luo, S. J. (2014). The genetics of brown coat color and white spotting in domestic yaks (Bos grunniens). Animal Genetics, 45, 652–659. doi:10.1111/age.12191
- Zhang, Q., Youji, M., Yong, Z., & Xingxu, Z. (2015). Identification of copy number variations in qinchuan cattle using BovineHD genotyping beadchip array. Molecular Genetics and Genomics: MGG, 290(1), 319–327. doi:10.1007/s00438-014-0923-4
- Zhang, R. (1989). China Yak. Lanzhou: Gansu Scientific, and Technology.
- Zhang, X., Wang, K., Wang, L., Yang, Y., Ni, Z., Xie, X., … Qiu, Q. (2016). Genome-wide patterns of copy number variation in the Chinese yak genome. BMC Genomics, 17, 379. doi:10.1186/s12864-016-2702-6