
Abstract
In this paper I describe more than 30 years of investigations of the autoinflammatory syndrome hyper-IgD syndrome (HIDS). In the first paper after the recognition of the syndrome published in 1984, we described the characteristics of this periodic fever syndrome. The hypotheses regarding the pathogenesis of the fever and the acute phase response in these patients prompted us to study interleukin-1 (IL-1), the cytokine formerly described as endogenous pyrogen and lymphocyte activating factor. Although we were unable to find elevated concentrations of IL-1 in the circulation, we discovered that white blood cells spontaneously produced elevated amounts of IL-1b. A major next discovery was the identification of the gene defect by us and others in 1999: quite unexpectedly the mevalonate kinase, an enzyme in the cholesterol synthesis pathway was found to be mutated. We were able to describe a founder effect and a phenotypic continuum with the classical mevalonate aciduria in the years to follow. A major step forward was the finding that recombinant interleukin-1 receptor antagonist (anakinra) was an effective treatment for the majority of patients. Thus, research over a period of three decades after the first recognition of the syndrome, has yielded much insight into the pathogenesis as well as an effective therapy for HIDS.
Introduction
In this legacy paper I will give an account of 30-year long travels between bedside and bench and vice versa because of a peculiar periodic fever syndrome, i.e., hyper-IgD syndrome. As the paper deals mainly with the results obtained by our group, this is a very biased literature review, with probably too little credits to the work of others.
Discovery of a new syndrome
As a young internist working in the Department of Infectious Diseases at the University Medical Center in Leiden in the early 1980's, I became intrigued by the febrile response of human beings. I had just finished my PhD on proliferation and differentiation of mononuclear phagocytes in long-term cultures under the guidance of Ralph van Furth, and was wondering how to proceed. I felt that the subject of my PhD thesis was not very well connected to clinical medicine. At that crossroad, we had admitted 2 unrelated male adolescents with unexplained fever attacks, patient M and patient S to our ward. Patient S had an older sister M-S with a similar picture, who I had seen at the outpatient clinic.
Because I was also building up an outpatient clinic for adolescents and adults with primary immunodeficiencies (such as agammaglobulinemia and phagocyte disorders), I had close contacts with the technicians and the laboratory staff at the Department of Immunohematology headed by Professor Jon van Rood. Once a week, there was a session on immunoglobulin measurements with Dr Jiri Radl (), who would scrutinize the immunoelectrophoresis patterns. At the time that patients M and S were admitted, Jiri showed the findings of their immunoelectropherograms, pointing to a distinct band, representing IgD, which was present in both. A week later I received a phone call from Riek Schuit, a technician of Dr Willy Hijmans. She had received bone marrow samples of patient M and of M-S, the sister of patient S. She asked me whether M and M-S were relatives, because of the remarkable resemblance of the immunofluorescence pattern of their bone-marrow specimens: both showed strong staining for cytoplasmic IgD and IgA. In her vast experience, Riek had never seen such a pattern before. I told her that these 2 were unrelated, and that she would receive a bone-marrow sample from S, the brother of M-S. At that point, I started to realize that we probably touched on a new syndrome, characterized by periodic fever and hyperimmunoglobulinemia D.
Figure 1. Jiri Radl, around 1984.

More patients
Within half a year, by sheer coincidence, another patient was referred to my outpatient clinic, a 16-year old girl with a life-long history of incapacitating fever attacks. Just like the other 3 patients, her attacks had started at the first childhood vaccinations, and she would have lymph node swelling and loose stools during the attacks. Some attacks were accompanied by a skin rash. I could hardly wait to see her immunoelectrophoresis pattern; it showed the most prominent IgD precipitation line we had seen up to then (). The quantification by single radial immunodiffusion showed a serum IgD concentration of 5300 IU/l, higher than what we had seen before.
Figure 2. Immunoelectrophoresis pattern showing IgD precipitation of serum IgD in a HIDS patient.
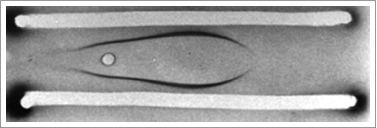
Soon after, 2 more patients were diagnosed. During the months to come we did a couple of obvious things. We investigated bone marrow of all patients, and did a series of additional immunological investigations. We also investigated whether patients with Familial Mediterranean Fever (FMF), the only established periodic fever syndrome at that time, had elevated IgD concentrations in their blood. With the exception of one Turkish girl, who showed a serum IgD of 177 IU/l (normal value < 150 IU/l), all FMF patients had low or undetectable IgD concentrations. We tried whether the drug that was shown to prevent attacks in FMF, colchicine would influence the attacks. There was no convincing preventive or therapeutic effect. Of course, we also speculated about the pathogenesis. Immune complexes at that time were a favorite mechanism of many immunologists, so we considered IgD complexes as an explanation for the pathogenesis.
That same year, Joost Oppenheim visited our Department and gave a seminar on lymphocyte activating factor (LAF), in which he highlighted the discovery of LAF as endogenous pyrogen,Citation1 and the new nomenclature in which this mediator was named interleukin-1 (IL-1). To me this was an eye opener: the immune system linked to the febrile response, this might well be a pathophysiological mechanism in our new syndrome. This was definitely a possibility to pursue.
Our first hyper-IgD paper
The paper was relatively easy to write. In first instance, we submitted it to New England Journal of Medicine. After a few weeks it came back judged by one reviewer. Naively, I had given the paper the title: 'Hyperimmunoglobulinaemia D and periodic fever, a syndrome resembling Familial Mediterranean Fever'. The reviewer stated: ‘this syndrome does not resemble FMF, since there is no clear serositis, there is lymphadenopathy and diarrhea’. This was enough for the Journal to reject the paper. Based on the review I decided to slightly rewrite it to emphasize the differences with FMF, and submitted it to Lancet as 'Hyperimmunoglobulinaemia D and periodic fever, a new syndrome'. Without any revision, it was immediately accepted and rapidly published.Citation2
I was convinced that we were the first to recognize this syndrome, because I had not been able to find similar descriptions in the literature written in English. Later it appeared that Anne-Marie Prieur and Claude Griscelli in Paris had published a paper in French in 1983, in which they described similar patients as a form of early-onset systemic juvenile arthritis.Citation3
Our Lancet paper was published in May 1984.Citation2 That same month Charles Dinarello published his interleukin-1 review in Reviews of Infectious Diseases.Citation4 I phoned him and asked whether I could visit Boston to discuss our papers, and whether he could teach me how to measure interleukin-1. I visited him in connection to a conference that I attended in Washington shortly thereafter. We had great discussions that day, he showed me the first Northern blots of IL-1β and we laid the basis for a friendship of decades.
Boston 1984 and 1987
In November 1984, I went to Boston where Charles' technician Gail LoPreste taught me the gel filtration of the plasma and the IL-1 bioassay using either D10 cells or mouse thymocytes. One of the first evenings in Boston, Charles invited me for a dinner at his house, where he had also invited his boss and mentor Sheldon Wolff with his family. Shelley, as he was generally called, had read my hyper-IgD paper and confessed that he had been the reviewer who destroyed the submission to the New England Journal. We became good friends.
Measuring IL-1 in plasma of patients with the assay did not really work. In samples from patients with an acute phase response (like in hyper-IgD syndrome), there was a tremendous amount of inhibiting molecules of about the same molecular weight as the 17 kD IL-1, leading to massive inhibition of the IL-1 bioassay.
However, my interest in IL-1 was kindled, and in 1987, I went to Charles in Boston for a sabbatical. This was a very productive period, because the conditions were incredible. Charles was (and is) a magnificent mentor (). Sheldon Wolff was there in the background. Recombinant IL-1 β, IL-1 α and TNF were just available and together with Stefan Endres, Gert Lonnemann and Joe Cannon, we developed the first specific radioimmunoassays to measure IL-1α, IL-1β and TNF.Citation5,6 We also worked on optimization of the assays for measuring IL-1β in plasma, using chloroform extraction and designed a whole blood cytokine assay.Citation7-9 In parallel I did a project on the effects of IL-1 on the innate defense to infection.Citation10
Figure 3. Charles Dinarello (right) and Jos van der Meer (cartoon by Jos van der Meer).

The year after my return from Boston, I was appointed as a full professor of internal medicine in Nijmegen.
Between 1984 and 1992, I had quite a lively correspondence with physicians from many parts of the world who had diagnosed patients with hyper-IgD syndrome. In that period a number of case reports were published as well. Remarkably, there were no American cases during that time frame.
Despite the fact that we had the sensitive and specific assay for IL-1β Citation11 we were not successful in finding measurable concentrations of this cytokine during attacks of patients with hyper-IgD syndrome.
Joost Drenth
In 1992, Joost Drenth MD, received a grant from the Dutch funding organization NWO to do a PhD on hyper-IgD syndrome. With great enthusiasm Joost started his project. The first things Joost and I did were contacting clinicians and scientists who had expressed their interest in hyper-IgD syndrome to establish the International hyper-IgD Study Group and building a registry of patients with the syndrome. This was the basis for international collaboration and further studies. Exactly 10 years after the Lancet paper, Joost together with the study group published the collective data on 50 patients with hyper-IgD syndrome in Medicine ().Citation12 He also described the cutaneous manifestations Citation13 and embarked on studies of cytokines and other inflammatory markers.Citation14-16 These studies revealed that there is a brisk acute phase response during the attack (as measured by CRP, α1-acid glycoprotein, and soluble PLA2), and a moderate rise in IL-6. No measurable increase in circulating IL-1β, IL-1α and a slight change in plasma TNF and interferon-γ was found during the attacks.Citation14,17 Anti-inflammatory mediators such as IL-1 receptor antagonist (IL-1ra) and soluble TNF receptors p55 and p75 were markedly elevated during the attacks.Citation14
Table 1. Symptoms and signs during attacks in patients with HID
More remarkable perhaps were the experiments on ex-vivo cytokine production. For the first time, it became clear that unstimulated white blood cells of asymptomatic hyper-IgD patients produced more IL-1β, IL-6 and TNF than controls.Citation14 Although unstimulated cells from FMF patients also produced more cytokines than controls, they produced less than those of hyper-IgD patients.Citation15 These data pointed to a pre-activated state of the monocytes in our patients, later also found in other periodic fever syndromes. During the attacks the unstimulated and stimulated cytokine production increased significantly.Citation14
Urinary neopterin was shown to be a reliable marker to assess attacks.Citation17
Taken together it was clear that there was an augmented cytokine and acute phase protein response in our patients, although we could not pinpoint the cytokine in the blood that was responsible for the systemic symptoms and signs (such as fever).
At that time we had lengthy discussions how to explain our inability to convincingly demonstrate the presence of major pyrogenic cytokines in the blood of febrile patients. Would it be only IL-6 doing the job as a circulating endogenous pyrogen and a mediator of the acute phase response? Could it be that IL-1β is only detectable in the circulation under extreme circumstances, such as sepsis?Citation11,18 Was it that our IL-1β assays were still lacking sensitivity? Was IL-1β hiding for our assays, e.g., by complexing with binding substances (a binding that was resistant to our chloroform extractionCitation7)? Was IL-1β only acting at the tissue level in a paracrine and autocrine fashion?
Another thing Joost and I did, was organizing the first international Hyper-IgD workshop in Nijmegen in 1995.Citation19 The workshop met with a lot of enthusiasm and it strengthened the international collaboration; here it was decided to adopt the abbreviation HIDS for the syndrome ().
Figure 4. First hyper-IgD working group meeting in Nijmegen, 1995 Joost Drenth in the middle of the first row, Jos van der Meer to the far right.

Search for the gene defect
The international collaboration had already facilitated the collection of genetic material of a number of families with patients with HIDS. The pedigrees were compatible with an autosomal recessive mode of inheritance. Following the work of Pras and Kastner et al in FMF,Citation20 we could demonstrate that in the case of HIDS, there was no linkage with chromosome 16p and neither with chromosomes 17q and 14q.Citation21
After Joost Drenth had cum laude defended his thesis (1996), and when the gene defect in FMF was discovered by the American Israeli consortiumCitation22 and the French group,Citation23 we decided to try and identify the gene for HIDS. We had been able to expand the number of informative families, and Joost obtained a grant from the Niels Stensen Foundation. For a genome-wide search, Joost went to Paris to collaborate with Marc Delpech and with Généthon. Within a couple of months, he was able to link HIDS to chromosome 12q and more precisely to the mevalonate kinase gene (MVK).Citation24 Independently, based on the serendipitous detection of mevalonate aciduria in a HIDS patient, Houten et al also discovered this gene defect. The papers were published back-to-back in Nature Genetics.Citation24,25 Interestingly, mevalonate aciduria (MIM 251170) was already known as a very serious inborn error of metabolism at that time. HIDS apparently represented a milder variant, with a residual mevalonate kinase activity of 5–15 %.
Nice to know the gene defect, but how to link a defect in the mevalonate pathway (the isoprenoid pathway) to an inflammatory, immunological disorder?
Anna Simon
In 1999, the young physician Anna Simon () started her residency in internal medicine in Nijmegen. She received a NWO grant for a training position as a clinical investigator in 2000 and started to work on HIDS. Her first accomplishment was to create a website HIDS.net and an electronic database for the registry. Next she was able to show that there was a founder effect for the most common MVK mutation in HIDS, V377I.Citation26 Another major accomplishment was the discovery by the neurologist Hubertus Kremer and Anna Simon of the phenotypic continuum between the classic mevalonate aciduria and HIDS: 5 mentally retarded adult patients suffering from cerebral ataxia were found to have MVK mutations and very low enzyme activity. Four of them suffered from fever episodes and had an elevated serum IgD.Citation27
Figure 5. Dr Anna Simon.

Another important finding was that approximately 25% patients in our registry had no MVK mutations. Anna found out that those without mutations (‘variant HIDS’) were most often sporadic, had a milder clinical picture, a somewhat later onset (i.e., not having the first attack at the first childhood vaccination), attacks of slightly longer duration (∼6.9 days versus 4.7 days) and lower serum IgD and IgA.Citation28 In hindsight one may question whether it is wise to use the diagnosis variant HIDS, because it is uncertain that these patients represent a single syndrome.
Regarding the pathophysiology of HIDS patients with MVK mutations a key question was whether the attacks were due to excess mevalonic acid (MA) or to a depletion of downstream products. Although I will come back to this issue at the end of this paper, our initial data were favoring the former mechanism. In-vitro we found evidence that MA was able to prime white cells for IL-1 and IL-6 production (A Simon, unpublished). More relevant perhaps were the results of a randomized controlled clinical trial, in which we found that simvastatin (which inhibits the formation of MA by inhibiting the reduction of HMG CoA) significantly decreased the number of febrile days in HIDS.Citation29 Others have suggested that the depletion of isoprenoids is responsible for the enhanced cytokine response through decreased geranylgeranylation of RAC1. Citation30
The concept of autoinflammatory syndromes
In 1999, also the genetic background of TRAPS, TNF receptor associated periodic syndrome, originally described as Familial Hibernian Fever, was elucidated/ In conjunction with that discovery, Daniel Kastner and co-workers coined the term ‘autoinflammatory syndromes’ to distinguish a group of disorders in which there was a ‘seemingly unprovoked inflammation without high-titer auto- or autoagressive T lymphocytes antibodies'.Citation31 Kastner et al later defined autoinflammatory syndromes as disorders with abnormally increased inflammation, mediated predominantly by the cells and the molecules of the innate immune system with a significant host predisposition.Citation32 It is clear that this definition distinguishes the autoinflammatory syndromes from autoimmune diseases. The term autoinflammatory syndromes has now largely replaced that of periodic fever syndromes.
25 years of HIDS
In November 2008, at the threshold of the 25th anniversary of the original description of HIDS, our PhD student Jeroen van der Hilst published a long-term follow up study of 103 HIDS patients with MVK mutations.Citation33 In this paper, it is reported that although the frequency of attacks in general decreases over the years, 50% of the patients over the age of 20, still experienced more than 6 attacks per year. Also it was found that HIDS interferes markedly with quality of life and hampers educational achievement and employment status. Long-term sequelae appeared to be very rare. AA amyloidosis, a major and relatively common threat in other autoinflammatory syndromes (such as untreated FMF, TRAPS) was found to be very rare in HIDS, despite a life-long duration of disease and frequent attacks with a brisk acute phase response. Jeroen van der Hilst did a series of experiments that made it plausible that blocking the isoprenoid pathway (as in HIDS) inhibits amyloidogenesis.Citation34
Therapy
Since the discovery of HIDS, we have been eagerly searching for an effective treatment. Pretty soon we found that colchicine, which is very effective in FMF, did not alleviate and prevent attacks. High dose steroids, however seem to have an effect in some patients,Citation33 but of course at the price of serious side effects. Based on preliminary positive experience with thalidomide in 2 HIDS patients, we performed a formal trial with this drug in 1999 – 2000; this study did not show a benefit.Citation35 The simvastatin trial, mentioned above met with a moderate effect.Citation29
Despite our inability to demonstrate that IL-1β was a circulating pyrogen in HIDS, we felt – based on the augmented ex-vivo IL-1β production mentioned aboveCitation14,15 – that IL-1β probably plays a major role in the pathogenesis. The way to demonstrate that would be an intervention directed toward IL-1. Already in 1995 (!) I sent a request to the manufacturer of Anakinra (recombinant IL-1 receptor antagonist) to provide the drug for compassionate use in HIDS. Although at that time the anakinra trial in sepsis patients was known to be negative,Citation36 the request was turned down without a thorough argumentation. Our experience with refusals like that kindled our initiative for a recent letter to Science Translational Medicine.Citation37 It took until 2005 before we could investigate the drug in HIDS.Citation38 In our initial studies with anakinra in HIDS, we used vaccination to provoke attacks; with the PhD student Evelien Bodar, we observed that anakinra was able to abort an attack, much more effectively than the TNF inhibitor etanercept.Citation38
At that time we had investigated whether the oral HDAC inhibitor ITF 2357 (givinostat), which inhibits a series of cytokines, would be effective in autoinflammatory disorders. The results of that pilot study did not demonstrate a sizable effect in HIDS.Citation39
In 2011 we could publish the results of a prospective observational study in which we showed decreased duration and severity of fever attacks with anakinra on demand.Citation40 Unlike in the studies in the cryopyrin-associated periodic syndromes,Citation41 anakinra was not effective in all patients, suggesting that HIDS is not a pure IL-1 disorder. More insight in the pathophysiology is needed to find new leads for treatment.
More pathophysiological studies
Returning to the question how to link a defect in the mevalonate pathway (the isoprenoid pathway) to an inflammatory, immunological disorder, the work of our former PhD student Monique Stoffels is relevant. She found that HIDS patients exhibit an enhanced response of the cytokines IL-1β, IL-1α, TNF and IL-6, specifically after stimulation of Toll-like receptors 4 and 2 and NOD2.Citation42 Within the activated cytokine network, it may well be that IL-1β is a major driver, since there is an enhanced ratio between active and inactive caspase-1, a major IL-1β activating enzyme.Citation42 In addition, experiments in vitro in the presence of IL-1 receptor antagonist showed decreased production of TNF and IL-6. Similar effects were seen when patients are treated with anakinra. However, Monique's finding that the IL-6-receptor blocking monoclonal antibody tocilizumab blocks the IL-1β response more than 5 fold and the observation mentioned above that not all HIDS patients respond to anakinra, point to a multi-cytokine disease.
Another link between the isoprenoid pathway and inflammation comes from recent reports by David Russell's group.Citation43,44 They found that 25-OH cholesterol is an inhibitor of IL-1β production. The production of 25-OH cholesterol as a metabolite of cholesterol is dependent on the enzyme cholesterol-25-hydroxylase (Ch25h) in monocytes. The transcription of this enzyme is under control of the type-1 interferon pathway.Citation43 Thus, it is conceivable that a deficiency of MVK leads to depletion of 25-OH cholesterol and hence to an enhanced IL-1β response.Citation45 It also might shed some light on the still enigmatic high concentrations of IgD and IgA in HIDS. Although not all patients with MVK mutations have a high serum IgD and IgA,Citation25,33 the majority of patients exhibits this immunoglobulin pattern, as does the MVK knockout mouse.Citation46 Interestingly, the Ch25h knockout mouse, also has high concentrations of IgA through an effect on B lymphocytes.Citation47 Whether these mice have high IgD is not known (D Russell, personal communication).
Final remarks
In this paper I have given a personal account of our 30 years research on HIDS. What started with a challenge at the bedside of 3 patients with periodic fever, and the remarkable laboratory findings of elevated serum IgD concentrations and a pecular bone-marrow immunofluoresence became a recognizable clinical syndrome. At the time of the first description, we speculated about the role of IL-1, at that time the only bonafide endogenous pyrogen.
The hunting for the gene yielded a peculiar link between the isoprenoid pathway (cholesterol synthesis pathway) and inflammatory mechanisms. To try and connect these different areas posed a new challenge. In the mean time the role of proinflammatory cytokines became more obvious, with a major, although not monopolistic role for IL-1.
Although anakinra is not universally effective in HIDS, it is beneficial in quite some patients, and for them it means that there is an effective treatment available now. The effect of the anti-IL-1β human monoclonal antibody (Canakinumab) still needs more study, like perhaps other treatments.
As I said in the introduction, the work on periodic fevers came from a bedside fascination for patients with fever of unknown origin (FUO). In Nijmegen I further explored this great clinical challenge with young internists like Elisabeth (Lilly) de Kleijn and Chantal Bleeker-Rovers.Citation48-50 With regard to the diagnostic approach we were able to make progress, especially using Fluoro desoxyglucose positron emission tomography (FDG-PET).Citation50
Over the past decades, I have felt very privileged for a number of reasons. As a clinician in the university hospitals in Leiden and Nijmegen, I felt challenged at the bedside, taking care of patients presented that with unclear clinical pictures. I have always tried to envisage what was going on in such patients in terms of pathophysiology, and tried to investigate that in our laboratory.
Secondly, the decision to get involved in the new and rapidly expanding cytokine field allowed us to explore the role of cytokines like IL-1 in host defense against infection and their role in non-infectious inflammation. This led to a highly productive research line that came to full bloom when Mihai Netea joined the group. He started as a summer student from Cluj, Rumania, in our laboratory; next he became a PhD student and a resident in internal medicine and infectious diseases. Currently he is a full professor of experimental medicine at our university. Of the large number of contributions to medical sciences of Mihai, one of the most appealing is without doubt the paradigm of trained immunity, which encompasses the induction of persistent activation of monocytes and macrophages through epigenetic mechanisms.Citation51-53
Satisfaction did not only come from taking care of ‘difficult’ patients, but also from mentoring young clinicians and clinical investigators, guiding them and stimulating them on their career path. I had the opportunity to be the mentor (‘promotor’) of more than 75 PhD candidates. Many of their PhD theses have contributed to the progress in medical sciences.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Rosenwasser LJ, et al. J Exp Med 1979; 150:709; PMID:314491; http://dx.doi.org/10.1084/jem.150.3.709
- Van der Meer JW, et al. Lancet 1984; 1:1087-90; PMID:6144826; http://dx.doi.org/10.1016/S0140-6736(84)92505-4
- Prieur AM, Griscelli C. Ann Pédiat 1983; 30:565-9; PMID:6638806
- Dinarello CA. Rev Infect Dis 1984; 6:51-95; PMID:6369481; http://dx.doi.org/10.1093/clinids/6.1.51
- Lonnemann G, et al. Lymphokine Res 1988; 7:75-84; PMID:3261380
- Van der Meer JW, et al. J Leukoc Biol 1988; 43:216-23; PMID:3257788
- Cannon JG, et al. Lymphokine Res 1988; 7:457-67; PMID:3145372
- Nerad JL, et al. J Leukoc Biol 1992; 52:687-92; PMID:1464740
- Endres S, et al. Clin Immunol Immunopathol 1988; 49:424-38; PMID:2461270; http://dx.doi.org/10.1016/0090-1229(88)90130-4
- Van der Meer JW, et al. Proc Natl Acad Sci U S A 1988; 85:1620-3; PMID:3125553; http://dx.doi.org/10.1073/pnas.85.5.1620
- Cannon JG, et al. J Infect Dis 1990; 161:79-84; PMID:2295861; http://dx.doi.org/10.1093/infdis/161.1.79
- Drenth JP, et al. Medicine 1994; 73:133-44; PMID:8190036; http://dx.doi.org/10.1097/00005792-199405000-00002
- Drenth JP, et al. Arch Dermatol 1994; 130:59-65; PMID:8285741; http://dx.doi.org/10.1001/archderm.1994.01690010063008
- Drenth JP, et al. Blood 1995; 85:3586-93; PMID:7780142
- Drenth JP, et al. J Immunol 1996; 157:400-4; PMID:8683144
- Havenaar EC, et al. Clin Immunol Immunopathol 1995; 76:279-84; PMID:7554449; http://dx.doi.org/10.1006/clin.1995.1126
- Drenth JP, et al. Eur J Clin Invest 1995; 25:683-6; PMID:7498243; http://dx.doi.org/10.1111/j.1365-2362.1995.tb01986.x
- Van Deuren M, et al. Blood 1997; 90:1101-8; PMID:9242541
- Drenth JP, Powell RJ, et al. Lancet 1995; 345:445-6; PMID:7853961; http://dx.doi.org/10.1016/S0140-6736(95)90420-4
- Pras E, Aksentijevich I, Gruberg L, Balow JE, Prosen L, Dean M, et al. N Engl J Med 1992; 326:1509-13; PMID:1579134; http://dx.doi.org/10.1056/NEJM199206043262301
- Drenth JP, et al. Hum Genet 1994; 94:616-20; PMID:7989036; http://dx.doi.org/10.1007/BF00206953
- The International FMF Consortium. Cell 1997; 90:797-807; PMID:9288758; http://dx.doi.org/10.1016/S0092-8674(00)80539-5
- French FMF consortium. Nat Genet 1997; 17:25-31; PMID:9288094; http://dx.doi.org/10.1038/ng0997-25
- Drenth JP, et al. Nat Genet 1999; 22:178-81; PMID:10369262; http://dx.doi.org/10.1038/9696
- Houten SM, et al. Nat Genet 1999; 22:175-7; PMID:10369261; http://dx.doi.org/10.1038/9691
- Simon A, et al. Am J Med 2003; 114:148-52; PMID:12586237; http://dx.doi.org/10.1016/S0002-9343(02)01429-8
- Simon A, et al. Neurology 2004 Mar 23; 62:994-7; PMID:15037710; http://dx.doi.org/10.1212/01.WNL.0000115390.33405.F7
- Simon A, et al. Ann Intern Med 2001; 135:338-43; PMID:11529697; http://dx.doi.org/10.7326/0003-4819-135-5-200109040-00010
- Simon A, et al. Clin Pharmacol Ther 2004; 75:476-83; PMID:15116060; http://dx.doi.org/10.1016/j.clpt.2004.01.012
- Kuijk LM, et al. Blood 2008 Nov 1; 112:3563-73; PMID:18684863; http://dx.doi.org/10.1182/blood-2008-03-144667
- Hull KM, et al. Curr Opin Rheumatol 2003; 15:61-9; PMID:12496512; http://dx.doi.org/10.1097/00002281-200301000-00011
- Kastner DL, et al. Cell 2010; 140:784-90; PMID:20303869; http://dx.doi.org/10.1016/j.cell.2010.03.002
- Van der Hilst JCH, et al. Medicine (Baltimore) 2008; 87:301-10; PMID:19011501; http://dx.doi.org/10.1097/MD.0b013e318190cfb7
- Van der Hilst JCH, et al. J Leukoc Biol 2008; 83:1295-9; PMID:18285405; http://dx.doi.org/10.1189/jlb.1107723
- Drenth JP, et al. J Pharmacol Exp Ther 2001; 298:1221-6; PMID:11504824
- Opal SM, et al. Crit Care Med 1997; 25:1115-24; PMID:9233735; http://dx.doi.org/10.1097/00003246-199707000-00010
- Van der Meer JWM, et al. Sci Transl Med 2014; 6:219; PMID:24431110; http://dx.doi.org/10.1126/scitranslmed.3007911
- Bodar EJ, et al. Neth J Med 2005; 63:260-4; PMID:16093577
- Bodar EJ, et al. Mol Med 2011; 17(5-6):363-8; PMID:21274502; http://dx.doi.org/10.2119/molmed.2011.00039
- Bodar EJ, et al. Ann Rheum Dis 2011; 70:2155-8; PMID:21859689; http://dx.doi.org/10.1136/ard.2011.149922
- Goldbach-Mansky R, et al. N Engl J Med 2006 Aug 10; 355:581-92; PMID:16899778; http://dx.doi.org/10.1056/NEJMoa055137
- Stoffels M, et al. Rheumatology (Oxford) 2015; 54:363-8; PMID:25173351
- Reboldi A, et al. Science 2014; 345:679-84; PMID:25104388; http://dx.doi.org/10.1126/science.1254790
- McDonald JG, et al. J Leukoc Biol 2010; 88:1071-2; PMID:21123296; http://dx.doi.org/10.1189/jlb.0710418
- Simon A. N Engl J Med 2014; 371:1933-5; PMID:25390746; http://dx.doi.org/10.1056/NEJMcibr1412016
- Hager EJ, et al. J Inherit Metab Dis 2012; 35:159-68; PMID:21607759; http://dx.doi.org/10.1007/s10545-011-9349-x
- Bauman DR, et al. Proc Natl Acad Sci U S A 2009; 106:16764-9; PMID:19805370; http://dx.doi.org/10.1073/pnas.0909142106
- De Kleijn EM, et al. Medicine 1997; 76:392-400; PMID:9413425; http://dx.doi.org/10.1097/00005792-199711000-00002
- De Kleijn EM, et al. Medicine 1997; 76:401-14; PMID:9413426; http://dx.doi.org/10.1097/00005792-199711000-00003
- Bleeker-Rovers CP, et al. Medicine 2007; 86:26-38; PMID:17220753; http://dx.doi.org/10.1097/MD.0b013e31802fe858
- Netea MG, et al. Cell Host Microbe 2011 May 19; 9:355-61; PMID:21575907; http://dx.doi.org/10.1016/j.chom.2011.04.006
- Saeed S, et al. Science 2014; 345:1251086; PMID:25258085; http://dx.doi.org/10.1126/science.1251086
- Cheng S-C, et al. Science 2014; 345:1250684; PMID:25258083; http://dx.doi.org/10.1126/science.1250684