
Abstract
The p53 tumor suppressor network plays a fundamental surveillance role in both homeostatic and adaptive cell biology. p53 is one of the most important barriers against malignant derailment of normal cells, orchestrating growth arrest, senescence, or cell death by linking many different pathways in response to genotoxic and non-genotoxic insults. p53 is the key broadband sensor for numerous cellular stresses such as DNA damage, hypoxia, oxidative stress, oncogenic signaling, and nucleolar stress. The crucial tumor suppressive and tissue homeostasis activity of p53 is its ability to activate cell death via multiple different pathways. A well-characterized biochemical function of p53 in the regulation of apoptosis is its role as a potent transcriptional regulator. p53 activates a panel of proapoptotic genes from the mitochondrial apoptotic and death receptor programs while repressing antiapoptotic Bcl2 family genes. In addition, over the last 10 y a growing body of evidence has also defined direct extranuclear non-transcriptional p53 activities within mitochondria-mediated cell death pathways that are based on p53 protein accumulation in cytosolic and mitochondrial compartments and protein-protein interactions. To date, transcription-independent p53-mediated cell death regulation has been described for apoptosis, necrosis, and autophagy. Because mitochondrial dysregulation is central to the development of a number of pathologic processes such as cancer and neurodegenerative and age-related diseases, understanding the direct roles of p53 protein in mitochondria has high translational impact and could facilitate the development of novel drug targets to combat these diseases. In this review we will mainly focus on mechanisms of p53-mediated transcription-independent cell death pathways at mitochondria.
Abbreviations
| AD | = | Alzheimer disease |
| CypD | = | cyclophilin D |
| IMM | = | inner mitochondrial membrane |
| MOMP | = | mitochondrial outer membrane permeabilization |
| mPT | = | mitochondrial permeability transition |
| PTP | = | permeability transition pore |
| ROS | = | reactive oxygen species |
p53-Mediated Apoptosis
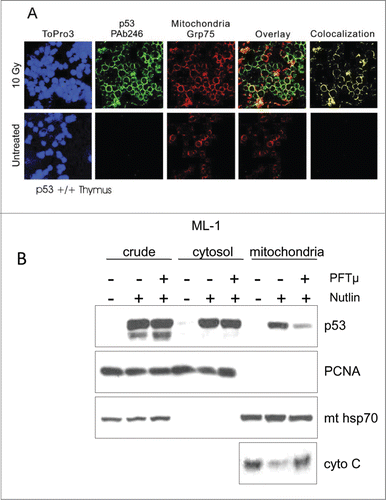
The notion that p53 may induce transcription-independent apoptosis in response to genotoxic stress was first suggested almost 20 y ago. In experiments with p53-null tumor cells that stably express a temperature-sensitive mutant p53 protein, Caelles et al. showed that at permissive temperatures the induction of apoptosis following DNA damage was independent of new RNA or protein synthesis.Citation1 These findings were subsequently confirmed in experiments of p53-null cells reconstituted with transcriptionally inactive truncation mutants of p53.Citation2,3 The mechanistic explanation for the transcription-independent apoptotic p53 activity came from pioneering studies that linked direct p53 protein action to the intrinsic mitochondrial apoptotic pathway.Citation4 We and others showed that, in response to stress, a fraction of induced p53 protein localizes to mitochondria during p-53 mediated apoptosis, where it behaves like a super BH3-only protein to induce mitochondrial outer membrane permeabilization (MOMP), physically interacting with and inhibiting antiapoptotic members (Bcl2, BclxL, Mcl-1) as well as activating proapoptotic members (Bak, Bax) of the Bcl2 family of MOMP regulators and thereby triggering p53-dependent apoptosis (see below).Citation4-6 Moreover, overexpression of antiapoptotic Bcl2 or BclxL abrogates stress signal-mediated mitochondrial p53 accumulation and subsequent apoptosis but not cell cycle arrest, suggesting a currently unexplored feedback signaling loop between p53 and apoptotic Bcl regulators.Citation4 Importantly, bypassing the nucleus by targeting p53 to mitochondria via fusions with an import leader peptide or the Bcl2-derived transmembrane domain is sufficient to induce apoptosis in p53-deficient cells.Citation4,7 This initial observation of stress-induced mitochondrial translocation of p53 was confirmed to occur in response to a multitude of apoptotic stimuli including genotoxic stress,Citation4 deregulated oncogenes,Citation8 hypoxia,Citation9 transcriptional blockade,Citation10 MDM2 inhibition (Fig. 1B), and heat stressCitation12 in cultured human and mouse normal, immortal, and cancer cells, and in response to gamma irradiation in vivoCitation13,14 in radiosensitive tissues such as thymus, spleen, and testis (Fig. 1A), but not in radioresistant tissues such as liver and kidney.Citation13,14
Figure 1. (A) Detection of stress-induced mitochondrial p53 accumulation in vivo by confocal immunofluorescence. Colocalization of p53 and mitochondrial marker protein Grp75 (mthsp70) in irradiated thymus. Normal mice were subjected to 10 Gy γIR or left untreated. After 2 h, tissue smears were prepared from thymi and double-stained with monoclonal anti-p53 antibody PAb 246 followed by FITC-goat anti-mouse antibody and a rabbit anti-Grp75 antibody followed by TRITC-donkey anti-rabbit antibody. Untreated cells exhibited no detectable staining with PAb 246. In contrast, all irradiated thymocytes exhibited prominent, finely punctated perinuclear ring staining, which exceeded the concomitant faint nuclear staining at this time point. Cytoplasmic p53 largely co-localized with mitochondria (yellow only). Nuclei were counterstained with ToPro3. (B) Inhibition of mitochondrial p53 translocation by PFTμ inhibits Mdm2 inhibitor Nutlin-induced apoptosis. Pre-treatment of ML-1 cells with PFTμ, a specific inhibitor of p53's association with mitochondria (see text), results in markedly reduced p53 translocation and reduced cytochrome C release in response to Nutlin treatment. ML-1 cells were pre-treated with 25 μM PFTμ for 2 h before adding 10 μM Nutlin for 12 h, followed by mitochondrial fractionation and immunoblotting as indicated. PCNA serves as loading control and nuclear contamination marker. mthsp70 is the loading control for mitochondria.
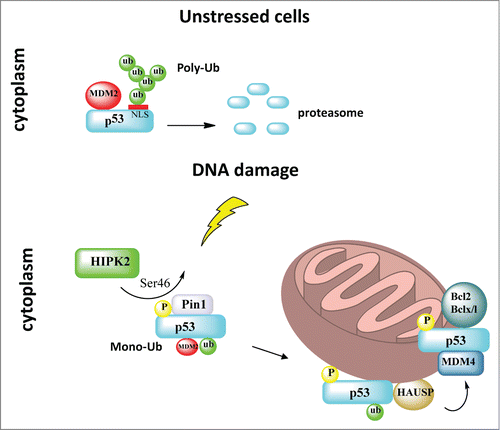
Pathways governing the translocation of p53 to mitochondria were found to be regulated by post-translational modifications. The first mechanistic insight came from work that demonstrated differential apoptotic potential of 2 common polymorphic variants of human p53 (Arg/Pro at codon 72), which perfectly correlated with their differential abilities to localize to mitochondria (with Arg72 being the more potent form for both killing and translocation).Citation15 The authors suggested that greater interaction of the p53 Arg72 variant with MDM2 might lead to enhanced ubiquitination of p53, with enhanced nuclear export and mitochondrial localization.Citation15 Subsequently, we mechanistically expanded on these observations and showed that the cytoplasm contains a separate distinct p53 pool, which is the major source for p53 translocation to mitochondria upon stress-induced stabilization. We found that the cytoplasmic p53 pool is subject to a binary switch from a fate of inactivation via polyubiquitination and degradation in unstressed cells, to stress-induced Mdm2-mediated monoubiquitination that masks the p53 nuclear localization sites, thereby locking it in the cytoplasm.Citation16 Multi-monoubiquitination of p53 on C-terminal lysine residues greatly promotes its translocation to mitochondria, thereby shifting the cell fate to apoptosis.Citation17 On the other hand, p53 does not require Mdm2 as a shuttle. Upon arrival at the mitochondrion, p53 undergoes rapid deubiquitination by resident mitochondrial HAUSP via a stress-induced mitochondrial p53-HAUSP complex to generate the apoptotically active non-ubiquitinated p53.Citation17 This mitochondrial translocation pathway was recently expanded by the identification of more components. Upstream, DNA damage-activated homeodomain interacting protein kinase 2 (HIPK2) induces phosphorylation of p53 at Ser46, which recruits Pin1 (a phospho-specific peptidylprolyl cis/trans isomerase that translates phosphorylation modifications into conformation changes of its target substrates) and triggers a phospho-dependent cis/trans isomerization of p53. This conformational change of cytoplasmic p53 reduces its affinity for Mdm2, resulting in an increase of monoubiquitinated p53 that undergoes mitochondrial translocation.Citation18-20 Sorrentino et al. showed that Pin1 promotes stress-induced localization of p53 to mitochondria in vitro and in vivo by stimulating p53 monoubiquitination.Citation18 Of note, Pin1 also plays an important activating role in stress kinase-mediated transcriptional activation of nuclear p53.Citation21 At the mitochondria, Ser46 phosphorylation of p53 mediates its binding to resident Mdm4 on the outer membrane, which in turn facilitates the interaction between p53 and Bcl2 to induce MOMP and mitochondrial apoptosis ().Citation22 Interestingly, the mitochondrial Ser46p53-Mdm4 axis might play a role in mediating a better prognosis in glioblastoma.Citation23
Figure 2. Mitochondrial translocation of p53. In unstressed cells the cytoplasmic pool of p53 is inactivated via MDM2-mediated polyubiquitination and degradation. DNA damage induces HIPK2-mediated p53 phosphorylation at Ser46, which recruits Pin1 and triggers a phospho-dependent cis/trans isomerization of p53. This conformational change of cytoplasmic p53 reduces its affinity for Mdm2, resulting in a switch from polyubiquitination to monoubiquitination that masks the p53 nuclear localization sites and promotes mitochondrial translocation of p53. Upon arrival at the mitochondria, p53 undergoes rapid deubiquitination by mitochondrial HAUSP via a stress-induced mitochondrial p53-HAUSP complex. Ser46-p53 phosphorylation at the mitochondrion also mediates its binding to resident Mdm4 on the outer membrane, which in turn facilitates interaction between p53 and Bcl2/BclxL to induce mitochondrial outer membrane permeabilization and mitochondrial apoptosis.
In addition to monoubiquitination of p53, Sykes et al. showed that a Lys120-acetylated form of p53 is enriched at the outer mitochondrial membrane after DNA damage. Lys120 acetylation is not required for p53 mitochondrial translocation, but rather enables p53 to displace the antiapoptotic Mcl-1 from the proapoptotic effector Bak, allowing Bak oligomerization and pore formation.Citation24 Consistent with these data, p53 acetylation at Lys320/Lys373/Lys382 is required for its transcription-independent functions in Bax activation and apoptosis in response to histone deacetylase inhibitors (HDACi).Citation25
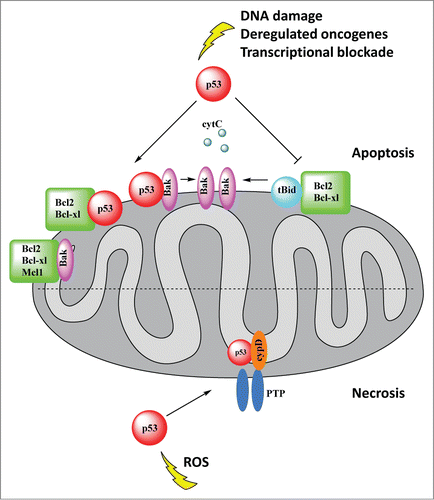
As briefly alluded to above, a major outcome of stress-induced mitochondrial translocation of p53 is the induction of transcription-independent apoptosis via the intrinsic mitochondrial pathway. This pathway depends on mitochondrial outer membrane permeabilization (MOMP), which is regulated by Bcl2 family members. Upon arrival at mitochondria, p53 physically interacts with Bcl2 family member proteins. The Bcl2 family of proteins is subdivided into 3 groups according to function as follows: (i) antiapoptotic BclxL, Bcl2, Bcl-w, and Mcl-1, which stabilize the outer membrane, (ii) the ultimate proapoptosis effectors Bax and Bak, which are able to form pores and/or associate with pore-forming proteins in the outer membrane; (iii) small BH3 domain-only proteins, which interact with both anti- and proapoptotic Bcl2 members to transmit the death signal and induce apoptosis (Puma, Noxa, Bad as ‘enablers’ on one hand; tBid and Bim as ‘activators’ on the other hand).Citation26 In response to stress, the cytoplasmic pool of p53 rapidly translocates to the surface of the outer mitochondrial membrane, where it behaves like a dual BH3-only protein, engaging in complexes with antiapoptotic (BclxL, Bcl2)Citation7,27,28 and/or proapoptotic (Bak)Citation6 members of the Bcl2 family. These interactions facilitate Bax and Bak homo-oligomerization, causing pore formation and permeabilization of the outer membrane and release of cytochrome c, Smac/Diablo, Htra2, and AIF, which activate the caspase cascade and ultimately induce apoptosis. Importantly, recombinant p53 induces MOMP and release of cytochrome c from isolated healthy mitochondria within minutes, confirming the direct effect of p53 on MOMPCitation5,7 (). Computational and mutational structure/function studies revealed that the DNA-binding interface of p53 is in direct contact with the BH4 domain and adjacent loops connecting α helices of BclxL (opposite the canonical BH123 binding pocket) to form the p53-BclxL complex.Citation7 This interaction was confirmed by several structural NMR studies.Citation27,29,30 Additionally, through direct binding to Bak, p53 is able to disrupt the complex between Bak and its antiapoptotic partner Mcl-1 at the outer membrane, leading to Bak oligomerization and MOMP ().Citation6 Notably, tumor-derived p53 missense mutants, although constitutively localized at the outer membrane as a result of their intrinsic cellular stabilization, are unable to bind to Bcl2 and trigger MOMP, indicating that missense mutants are ‘double hit’ loss-of-function mutants for apoptosisCitation27 and suggesting that the transcription-independent direct mitochondrial apoptosis program might participate in tumor suppression.
Figure 3. p53 mediates 2 programmed cell death pathways, apoptosis and necrosis, directly at the mitochondria. Top, Apoptosis: Upon arrival at the mitochondria, p53 physically interacts with antiapoptotic Bcl2 family members and proapoptotic Bak, facilitating Bak oligomerization. This in turn causes pore formation and permeabilization of the outer membrane and release of cytochrome c and other apoptogenic factors. The inhibitory interaction of p53 with BclxL/Bcl2 also liberates proapoptotic tBid from its inhibitory complex with BclxL/Bcl2. p53 interaction with Bak induces a conformational change in Bak, promoting Bak oligomerization and pore formation. Cytochrome c activates the caspase cascade and ultimately apoptosis. Bottom, Necrosis: In response to oxidative stress, p53 accumulates in the mitochondrial matrix and triggers opening of the mitochondrial permeability transition pore (mPTP) by physical interaction with its critical regulator cyclophilin D (CypD), which leads to necrosis.
An extension of this mitochondrial outer membrane-based p53 program that integrates concomitant transcription by nuclear p53 was proposed by the Green laboratory. In this model, p53 in unstressed cells is sequestered in the cytoplasm by soluble BclxL. Upon genotoxic stress, nuclear p53 transcriptionally induces Puma, which binds to cytosolic BclxL, thereby liberating p53. p53 can then bind and activate monomeric Bax in the cytoplasm via transient formation of a ‘hit-and-run’ complex, causing Bax homo-oligomerization and translocation to the outer membrane.Citation5,26 This mechanism appears to be cell-context dependent because the p53 transcription-independent direct mitochondrial apoptotic program also occurs in Puma−/− cells.Citation31
The major physiological outcome of p53WT-mediated MOMP is cytochrome c release, which leads to activation of the executioner caspases 3, 6, and 7 via the apoptosome.Citation32 Caspase-3, the key executioner caspase, is localized both in the cytosol and at the inner and outer mitochondrial membrane. Frank et al. identified procaspase-3 as a mitochondrial p53-interacting protein. Interestingly, tumor-derived mutant p53 proteins also bind procaspase-3 but interfere with its ability to be proteolytically activated by caspase-9. These data suggest that p53 mutants may be selected for in tumor cells because of their ability to bind and inhibit activation of caspase-3 and therefore block apoptosis.Citation33
Mitochondrial p53-Mediated Necrosis
The role of p53 at mitochondria in determining cell fate is not restricted to activation of apoptosis. We recently identified a transcription-independent direct mitochondrial matrix-based program of p53 that induces necrosis, a major form of cell death that is distinct from apoptosis in microscopic structure and biochemical and molecular features.Citation11 Necrosis is irreversible tissue destruction as a result of bioenergetic failure, and is central to the ischemia/reperfusion injury and oxidative damage that occurs in stroke and organ infarctions. The fundamental difference between necrosis and apoptosis is the rapid loss of cellular membrane potential as a result of energy depletion and ion pump/channel failure, leading to hydropic swelling, organellar rupture, and cytolysis with extracellular spilling of cellular content. The major mediators of necrosis are excess levels of cytosolic Ca2+ and reactive oxygen species (ROS). Rather than being a passive event, ischemic necrosis has emerged as a controlled cell death process. In contrast to apoptosis, necrosis induces an inflammatory response to stimulate tissue repair by selectively releasing immunostimulatory factors such as HMGB1 and HDGF from dying cells.Citation34 During ischemic injury, ROS and calcium levels sharply increase in cells and in the mitochondrial matrix. This increase triggers programmed necrosis through sudden opening of the mitochondrial permeability transition pore (mPTP), a normally impermeable non-selective water, ion, and small solute channel in the inner mitochondrial membrane (IMM) that is uniquely controlled by cyclophilin D (CypD). CypD, which is located in the matrix, is the essential regulator of PTP opening and the only genetically proven indispensable PTP component.Citation35
CypD is the sole mitochondrial member of the cyclophilin family of peptidyl prolyl cis-trans isomerases, which have a crucial role in protein folding and adaptive conformational changes. Recent studies with 4 independent knockout mouse strains definitively identified CypD as the key regulator of the mPTP pore at the inner membrane, opening of which leads to necrotic cell death.Citation36 CypD null mice are remarkably protected from ischemia/reperfusion-induced myocardial infacrction and stroke in vivo. Conversely, cyclophilin D-overexpressing mice show mitochondrial swelling and spontaneous cell death.Citation36-39 In agreement with these findings, mitochondria isolated from liver, heart, and brain of CypD null mice are resistant to calcium spike-induced mitochondrial swelling and permeability transition in vitro. Moreover, primary hepatocytes and fibroblasts isolated from CypD null mice are largely protected from necrotic death induced by Ca2+ overload and oxidative stress. Treatment with cyclosporine A, a highly specific CypD catalytic site inhibitor, reduces ischemia/reperfusion injury in the brain and heart.Citation37-39 Importantly, CypD-deficient cells die normally by apoptosis, emphasizing the existence of 2 functionally distinct mitochondrial death pathways.Citation37-40
In healthy cells, pumping H+ out of the matrix creates the proton gradient ΔΨm across the IMM that is necessary to maintain mitochondrial respiration and ATP production. In unstressed cells the PTP is closed and the IMM is impermeable to ions. Upon oxidative stress, sudden PTP opening causes massive ion influx that dissipates ΔΨm and shuts down oxidative phosphorylation and ATP production. This is called the mitochondrial permeability transition (mPT). Concomitant water influx causes matrix swelling, rupture of the rigid outer mitochondrial membrane, and release of all sequestered cell death factors. mPT is triggered by mitochondrial matrix sequestration of the high levels of cytosolic Ca2+ and ROS generated during oxidative damage. Clinically, CypD-triggered PTP opening and mPT are the driving pathophysiological forces behind cerebral stroke, myocardial infarction, and other vascular catastrophes that are leading causes of death. The mechanism by which CypD becomes activated to induce mPT remains unclear but is known to depend on its prolyl isomerase activity. Cyclosporine A (CsA) potently and specifically prevents mPT by binding to CypD, thus inhibiting its isomerase activity and displacing it from the PTP.Citation35
The first report that linked p53 with programmed necrosis showed that upon DNA damage Bax/Bak double knockout (DKO) mouse embryonic fibroblasts (MEFs) undergo necrosis, which is largely controlled by p53-mediated transcription of cathepsin Q in cooperation with DNA damage-induced ROS.Citation41 Interestingly, this transcriptional p53-cathepsin Q axis only plays a minor role in the corresponding necrosis in wild-type cells. In contrast, H2O2-induced necrosis in DKO MEFs is completely transcription-independent.Citation41 We subsequently found that, in contrast to the prototypical MOMP-inducer tBid, purified p53 protein has efficacy beyond activating the Bax/Bak lipid pore at the outer membrane. Recombinant p53 is able to rapidly release inner membrane-tethered apoptosis inducing factor (AIF) from healthy isolated Bax/Bak DKO mitochondria, whereas tBid fails to do so. Following up on this observation, we identified a novel transcription-independent function of p53 in the activation of necrotic cell death.Citation11 In response to oxidative stress and ischemia, p53 protein accumulates in the mitochondrial matrix and triggers PTP pore opening, mPT, and necrosis by physical interaction with the critical PTP regulator CypD (). This p53 action is transcription-independent (refractory to pifithrin α and α-amanitin) and is inhibited by CsA or by genetic CypD deletion or knockdown. Similarly, p53 causes structural alterations of the PTP pore, but tBid does not. Knockout and knockdown experiments of each partner in mouse and human cells demonstrated that oxidative stress induces PTP opening, mPT, collapse of ΔΨm, and necrosis that all concomitantly depend on the presence of p53 and CypD, and thus are interdependent. Most intriguingly, in an ischemia/reperfusion injury stroke model in mice, a robust p53-CypD complex is formed during necrosis in WT brain tissue. Conversely, genetic reduction of p53 levels (p53+/− mice) or CsA-pretreatment of p53WT mice is associated with effective stroke protection and a concomitant lack of pathologic p53-CypD complex formation.Citation11 Overall, these data imply a pathophysiologic role of the mitochondrial p53-CypD axis in ischemic stroke. Prior to our mechanistic study,Citation11 an intriguing study using rat neonatal brain ischemia to model perinatal hypoxia/asphyxia-induced brain damage had shown that blocking p53 translocation to mitochondria with pifithrin-μ (PFT-μ) suppressed oxidative stress and strongly reduced tissue necrosis and apoptosis. Thus PFT-μ although not exclusively restricted to inhibiting mitochondrial p53 action, was remarkably neuroprotective, with improved functional outcome and greatly decreased lesion size.Citation42 Of note, the neuroprotective effect of inhibiting only p53 transcriptional activity by PFT-α was significantly smaller and did not involve reduced oxidative stress.Citation42 Together, these studies imply that the direct mitochondrial p53-based necrotic and apoptotic machineries are major pathophysiologic effectors of ischemic brain damage, and that their interception might have promising therapeutic potential.
Aside from p53, very few extramitochondrial proteins are known to bind CypD and trigger pathologic PTP opening. A compelling analogy to p53, albeit acting chronically, is the highly toxic soluble intracellular form of β amyloid peptide (Aβ), a key protein in Alzheimer disease (AD), that progressively accumulates in brain mitochondria of patients with AD and in AD transgenic mice. In AD, Aβ forms a critical complex with CypD that promotes PTP opening, thereby boosting Ca2+-induced mitochondrial damage and neuronal injury. Inhibiting PTP or ablating CypD in AD mice renders their cortical brain mitochondria resistant to PTP opening and improves their cognitive functions.Citation43
The mitochondrial p53-CypD-PTP signaling pathway of necrosisCitation11 has since been independently confirmed and expanded to cover additional necrotic stimuli. As a model of oxidative stress-induced necrotic cell death of neurons, SH-SY5Y neuroblastoma cells were subjected to ischemia/reperfusion conditions via oxygen glucose deprivation/re-oxygenation. The ensuing necrosis was associated with mitochondrial p53 translocation and CypD complex formation, whereas disruption of the complex (by CsA, CypD knockdown, or p53 deletion/knockdown) inhibited the mPTP-mediated neuronal necrosis.Citation44 In addition, prolonged glucocorticoid exposure-mediated osteoporosis/osteonecrosis was modeled in cultured murine osteoblasts. Dexamethasone (Dex) induced necrotic death via mPTP opening, as indicated by its suppression by CsA, Sanglifehrin A (another specific CypD inhibitor), or CypD knockdown. Dex-induced events were associated with phosphorylation of p53 and its translocation to mitochondria, where it formed a complex with CypD. Moreover, glucocorticoid receptor knockdown or p53 inhibition (by shRNA silencing) suppressed Dex-induced p53-CypD complexation and necrosis.Citation45 Furthermore, chemotherapy-induced necrosis in p53WT cancer cells also involves the p53-CypD-mPTP axis. Loss-of-function analysis showed that cisplatin-induced necrosis of pancreatic cancer lines AsPC-1 and Capan-2Citation46 and doxorubicin-induced necrosis of lung cancer cells A459 require the mitochondrial p53-CypD signaling axis.Citation47
In summary, in addition to the well-characterized mitochondrial p53 program of apoptosis, a spectrum of stress stimuli including hypoxia, oxidative stress, DNA damage, and excess glucocorticoid induce a distinct mitochondrial p53-CypD program of necrosis. The decision of whether to engage one and/or the other program appears to depend on cell type and the nature or magnitude of the stress. Additional necrotic stimuli might exist and may also be context dependent. In addition, a recent study reported another type of p53-dependent, Bax/Bak-independent necrotic death in MEFs and human tumor cells. Although the precise mechanism remains to be elucidated, this process involves DNA damage induced by very high doses of H2O2 and is mediated by PARP-1, suggesting that in this scenario p53 can modulate the enzymatic activity of PARP-1. Interestingly, this form of cell death is not inhibited by CsA and minimally affected by CypD knockout, and is thus probably PTP-independent.Citation48 Furthermore, necrotic signaling also uses p53-independent pathways depending on the insult; for example, DNA alkylating agents induce necrosis through PARP and the TNFR/RIP1 pathways.Citation34
Pathophysiology of Mitochondrial p53 Death Pathways and Clinical Implications
In addition to the CNS ischemia studies discussed above,Citation11,42,44 there is growing evidence supporting a critical role of mitochondrial p53 in death signaling upon ischemic cerebral injury. Using microdissected hippocampal CA1 tissue instead of whole brain in a transient global cerebral ischemia model in rats, Endo et al. showed that mitochondrial p53 accumulation in hypoxia-sensitive hCA1 neurons strongly correlates with (i) formation of a p53-BclxL complex in isolated CA1 mitochondria, (ii) cytochrome c release, and (iii) neuronal death within 24 h. The pan-p53 small molecule inhibitor PFTα suppressed p53 transcription and p53 mitochondrial translocation and blocked neuronal cell death by 70% in vivo. Conversely, mitochondrial p53 accumulation was not detected in hypoxia-resistant CA3 neurons.Citation28,49
Proper mitochondrial length is critical for optimal mitochondrial function, and its dysregulation plays a causal role in neurologic and other pathologies. p53 directly influences the regulation of mitochondrial dynamics by modulating the expression and activity of fission and fusion proteins, dynamin-related GTPases that include Drp1 (the main fission protein) in a transcription-dependent and -independent manner. p53 transcriptionally induces mitofusin-2 (a fusion protein) and Drp1 under apoptotic conditions in HepG2 cells and cardiomyocytes, respectively.Citation50,51 Moreover, Huntington's disease (HD), which is caused by mutated Huntingtin protein (mtHtt), exhibits several cellular dysfunctions including excessive mitochondrial fission caused by hyperactivation of Drp1 at mitochondria. Accumulation of p53 on mitochondria is observed in animal and cell culture models of HD, and is associated with mitochondrial depolarization and mitochondrial complex IV inactivity.Citation52,53 An elegant study using mtHtt knock-in mouse neurons and human HD-iPS cell-derived striatal neurons implicates direct mitochondrial p53-Drp1 complex formation as a causal event for excessive mitochondrial fragmentation and necrosis in HD neurons. Cytoplasmic p53 binds to Drp1 and induces co-accumulation of Drp1 and p53 at mitochondria, leading to Drp1 hyperactivation and subsequent excessive mitochondrial fission and dysfunction (ROS production, loss of ΔΨm) and neuronal necrosis. Suppression of p53 (by siRNA), but not additional simultaneous Drp1 inhibition, effectively blocks all of these events.Citation54 In addition to Huntington's disease, a link to p53-Drp1 also exists in oxidative stress-induced necrosis. Using cell models, Guo et al. recently showed that under conditions of oxidative stress Drp1 stabilizes p53 and is required for p53 mitochondrial translocation through the induction of p53 monoubiquitination. Binding of Drp1 to p53 is associated with mitochondrial damage and necrosis. Most notably, inhibition of Drp1 hyperactivation by a peptide inhibitor (P110) blocks p53 mitochondrial accumulation and strongly reduces brain infarction in a rat ischemia reperfusion stroke model.Citation54
Another important translational aspect of mitochondrial p53 function comes from pioneering work of the Gudkov laboratory.Citation13 p53-mediated apoptosis in normal rapidly dividing radiosensitive and chemosensitive organs (e.g., the gastrointestinal lining and bone marrow) is a major cause of severe side effects during cancer treatment by radiation or chemotherapy, and often limits their efficacy. Studies to determine the effect of the apoptotic mitochondrial p53 program on radiosensitivity of normal tissue identified the small molecule pifithrin μ (PFTμ), which inhibits p53 accumulation at mitochondria by reducing its affinity to antiapoptotic BclxL and Bcl2 without affecting p53-dependent transactivation. Remarkably, a single intraperitoneal injection of PFTμ 30 minutes prior to treatment with 8-9 Gy of gamma radiation rescued otherwise lethally irradiated mice from bone marrow failure and strongly reduced radiation-induced thymocyte death in vitro.Citation13 Thus, the direct mitochondrial p53 program might be at least partly responsible for the adverse side effects that occur in sensitive normal tissues during cancer treatment and in acute radiation accidents. In vivo radioprotection through inhibition of the mitochondrial p53 program with PFTμ is an encouraging possible approach to suppressing the toxic side effects of chemo/radiotherapy and increasing the efficacy of anticancer treatments.
An interesting mouse model demonstrating the pathophysiological role of mitochondrial p53 function in liver was described by the George group.Citation55 The liver is generally refractory to p53-mediated apoptosis after genotoxic stress, in part due to transcriptional induction of the prosurvival factor IGFBP1 in the liver by stress-activated p53. IGFBP1 translocates to mitochondria, where it binds to proapoptotic Bak and prevents Bak activation, thus inhibiting apoptosis induction. In contrast, the liver of IGFBP1−/− mice exhibits spontaneous apoptosis associated with mitochondrial p53 accumulation and Bak oligomerization. On the other hand, blockade of p53 mitochondrial translocation by PFTμ in WT mice significantly reduces cisplatin-induced liver cell apoptosis. Along the same line, fatal liver ‘necrosis’ that occurs after α-amanitin poisoning from the highly toxic mushroom Amanitis phalloides is caused by the direct mitochondrial p53 death program. α-amanitin is an extremely potent transcriptional inhibitor (thus excluding any participation of the nuclear transcriptional function of p53) that was shown to induce p53-dependent apoptosis (and probably also necrosis, although this was not investigated) via mitochondrial p53 translocation and p53-dependent MOMP in liver and other cell types.Citation10,55 Accordingly, p53−/− and Bak−/− mice are largely protected from α-amanitin-induced liver damage, whereas wild-type mice undergo organ destruction.Citation55
In summary, the pathophysiological significance of proapoptotic and pronecrotic functions of mitochondrial p53 has been demonstrated in a number of compelling in vivo studies. Such studies have stimulated exploration into the translational potential of targeted interception of these pathways.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Caelles C, Helmberg A, Karin M. p53-dependent apoptosis in the absence of transcriptional activation of p53-target genes. Nature 1994; 370(6486):220-3; PMID:8028670; http://dx.doi.org/10.1038/370220a0
- Wagner AJ, Kokontis JM, Hay N. Myc-mediated apoptosis requires wild-type p53 in a manner independent of cell cycle arrest and the ability of p53 to induce p21waf1cip1. Genes Dev 1994; 8(23):2817-30; PMID:7995520; http://dx.doi.org/10.1101/gad.8.23.2817
- Haupt Y, Rowan S, Shaulian E, Vousden KH, Oren M. Induction of apoptosis in HeLa cells by trans-activation-deficient p53. Genes Dev 1995; 9(17):2170-83; PMID:7657168; http://dx.doi.org/10.1101/gad.9.17.2170
- Marchenko ND, Zaika A, Moll UM. Death signal-induced localization of p53 protein to mitochondria. A potential role in apoptotic signaling. J Biol Chem 2000; 275(21):16202-12; PMID:10821866; http://dx.doi.org/10.1074/jbc.275.21.16202
- Chipuk JE, Kuwana T, Bouchier-Hayes L, Droin NM, Newmeyer DD, Schuler M, Green DR. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 2004; 303(5660):1010-4; PMID:14963330; http://dx.doi.org/10.1126/science.1092734
- Leu JI, Dumont P, Hafey M, Murphy ME, George DL. Mitochondrial p53 activates Bak and causes disruption of a Bak-Mcl1 complex. Nat Cell Biol 2004; 6(5):443-50; PMID:15077116; http://dx.doi.org/10.1038/ncb1123
- Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P, Moll UM. p53 has a direct apoptogenic role at the mitochondria. Mol Cell 2003; 11(3):577-90; PMID:12667443; http://dx.doi.org/10.1016/S1097-2765(03)00050-9
- Nemajerova A, Wolff S, Petrenko O, Moll UM. Viral and cellular oncogenes induce rapid mitochondrial translocation of p53 in primary epithelial and endothelial cells early in apoptosis. FEBS Lett 2005; 579(27):6079-83; PMID:16226255; http://dx.doi.org/10.1016/j.febslet.2005.09.074
- Sansome C, Zaika A, Marchenko ND, Moll UM. Hypoxia death stimulus induces translocation of p53 protein to mitochondria. Detection by immunofluorescence on whole cells. FEBS Lett 2001; 488(3):110-5; PMID:11163756; http://dx.doi.org/10.1016/S0014-5793(00)02368-1
- Arima Y, Nitta M, Kuninaka S, Zhang D, Fujiwara T, Taya Y, Nakao M, Saya H. Transcriptional blockade induces p53-dependent apoptosis associated with translocation of p53 to mitochondria. J Biol Chem 2005; 280(19):19166-76; PMID:15753095; http://dx.doi.org/10.1074/jbc.M410691200
- Vaseva AV, Marchenko ND, Ji K, Tsirka SE, Holzmann S, Moll UM. p53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell 2012; 149(7):1536-48; PMID:22726440; http://dx.doi.org/10.1016/j.cell.2012.05.014
- Gu ZT, Wang H, Li L, Liu YS, Deng XB, Huo SF, Yuan FF, Liu ZF, Tong HS, Su L. Heat stress induces apoptosis through transcription-independent p53-mediated mitochondrial pathways in human umbilical vein endothelial cell. Sci Rep 2014; 4: 4469; PMID:24667845
- Strom E, Sathe S, Komarov PG, Chernova OB, Pavlovska I, Shyshynova I, Bosykh DA, Burdelya LG, Macklis RM, Skaliter R. Small-molecule inhibitor of p53 binding to mitochondria protects mice from gamma radiation. Nat Chem Biol 2006; 2(9):474-9; PMID:16862141; http://dx.doi.org/10.1038/nchembio809
- Erster S, Mihara M, Kim RH, Petrenko O, Moll UM. In vivo mitochondrial p53 translocation triggers a rapid first wave of cell death in response to DNA damage that can precede p53 target gene activation. Mol Cell Biol 2004; 24(15):6728-41; PMID:15254240; http://dx.doi.org/10.1128/MCB.24.15.6728-6741.2004
- Dumont P, Leu JI, Della Pietra AC, 3rd, George DL, Murphy M. The codon 72 polymorphic variants of p53 have markedly different apoptotic potential. Nat Genet 2003; 33(3):357-65; PMID:12567188; http://dx.doi.org/10.1038/ng1093
- Marchenko ND, Hanel W, Li D, Becker K, Reich N, Moll UM. Stress-mediated nuclear stabilization of p53 is regulated by ubiquitination and importin-alpha3 binding. Cell Death Differ 2010; 17(2):255-67; PMID:19927155; http://dx.doi.org/10.1038/cdd.2009.173
- Marchenko ND, Wolff S, Erster S, Becker K, Moll UM. Monoubiquitylation promotes mitochondrial p53 translocation. EMBO J 2007; 26(4):923-34; PMID:17268548; http://dx.doi.org/10.1038/sj.emboj.7601560
- Sorrentino G, Mioni M, Giorgi C, Ruggeri N, Pinton P, Moll U, Mantovani F, Del Sal G. The prolyl-isomerase Pin1 activates the mitochondrial death program of p53. Cell Death Differ 2013; 20(2):198-208; PMID:22935610; http://dx.doi.org/10.1038/cdd.2012.112
- D’Orazi G, Cecchinelli B, Bruno T, Manni I, Higashimoto Y, Saito S, Gostissa M, Coen S, Marchetti A, Del Sal G, et al. Homeodomain-interacting protein kinase-2 phosphorylates p53 at Ser 46 and mediates apoptosis. Nat Cell Biol 2002; 4(1):11-9; PMID:11780126; http://dx.doi.org/10.1038/ncb714
- Hofmann TG, Moller A, Sirma H, Zentgraf H, Taya Y, Droge W, Will H, Schmitz ML. Regulation of p53 activity by its interaction with homeodomain-interacting protein kinase-2. Nat Cell Biol 2002; 4(1):1-10; PMID:11740489; http://dx.doi.org/10.1038/ncb715
- Zacchi P, Gostissa M, Uchida T, Salvagno C, Avolio F, Volinia S, Ronai Z, Blandino G, Schneider C, Del Sal G. The prolyl isomerase Pin1 reveals a mechanism to control p53 functions after genotoxic insults. Nature 2002; 419(6909):853-7; PMID:12397362; http://dx.doi.org/10.1038/nature01120
- Mancini F, Di Conza G, Pellegrino M, Rinaldo C, Prodosmo A, Giglio S, D'Agnano I, Florenzano F, Felicioni L, Buttitta F, et al. MDM4 (MDMX) localizes at the mitochondria and facilitates the p53-mediated intrinsic-apoptotic pathway. EMBO J 2009; 28(13):1926-39; PMID:19521340; http://dx.doi.org/10.1038/emboj.2009.154
- Wang CL, Wang JY, Liu ZY, Ma XM, Wang XW, Jin H, Zhang XP, Fu D, Hou LJ, Lu YC. Ubiquitin-specific protease 2a stabilizes MDM4 and facilitates the p53-mediated intrinsic apoptotic pathway in glioblastoma. Carcinogenesis 2014; 35(7):1500-9; PMID:24445145; http://dx.doi.org/10.1093/carcin/bgu015
- Sykes SM, Mellert HS, Holbert MA, Li K, Marmorstein R, Lane WS, McMahon SB. Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Mol Cell 2006; 24(6):841-51; PMID:17189187; http://dx.doi.org/10.1016/j.molcel.2006.11.026
- Yamaguchi H, Woods NT, Piluso LG, Lee HH, Chen J, Bhalla KN, Monteiro A, Liu X, Hung MC, Wang HG. p53 acetylation is crucial for its transcription-independent proapoptotic functions. J Biol Chem 2009; 284(17):11171-83; PMID:19265193; http://dx.doi.org/10.1074/jbc.M809268200
- Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR. The BCL-2 family reunion. Mol Cell 2010; 37(3):299-310; PMID:20159550; http://dx.doi.org/10.1016/j.molcel.2010.01.025
- Tomita Y, Marchenko N, Erster S, Nemajerova A, Dehner A, Klein C, Pan H, Kessler H, Pancoska P, Moll UM. WT p53, but not tumor-derived mutants, bind to Bcl2 via the DNA binding domain and induce mitochondrial permeabilization. J Biol Chem 2006; 281(13):8600-6; PMID:16443602; http://dx.doi.org/10.1074/jbc.M507611200
- Endo H, Kamada H, Nito C, Nishi T, Chan PH. Mitochondrial translocation of p53 mediates release of cytochrome c and hippocampal CA1 neuronal death after transient global cerebral ischemia in rats. J Neurosci: Official J Soc Neurosci 2006; 26(30):7974-83; PMID:16870742; http://dx.doi.org/10.1523/JNEUROSCI.0897-06.2006
- Petros AM, Gunasekera A, Xu N, Olejniczak ET, Fesik SW. Defining the p53 DNA-binding domainBcl-x(L)-binding interface using NMR. FEBS Lett 2004; 559(1-3):171-4; PMID:14960327; http://dx.doi.org/10.1016/S0014-5793(04)00059-6
- Sot B, Freund SM, Fersht AR. Comparative biophysical characterization of p53 with the apoptotic BAK and the anti-apoptotic BCL-xL. J Biol Chem 2007; 282(40):29193-200; PMID:17699158; http://dx.doi.org/10.1074/jbc.M705544200
- Wolff S, Erster S, Palacios G, Moll UM. p53's mitochondrial translocation and MOMP action is independent of Puma and Bax and severely disrupts mitochondrial membrane integrity. Cell Res 2008; 18(7):733-44; PMID:18504456; http://dx.doi.org/10.1038/cr.2008.62
- Taylor RC, Cullen SP, Martin SJ. Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol 2008; 9(3):231-41; PMID:18073771; http://dx.doi.org/10.1038/nrm2312
- Frank AK, Pietsch EC, Dumont P, Tao J, Murphy ME. Wild-type and mutant p53 proteins interact with mitochondrial caspase-3. Cancer Biol Ther 2011; 11(8):740-5; PMID:21307660; http://dx.doi.org/10.4161/cbt.11.8.14906
- Zong WX, Thompson CB. Necrotic death as a cell fate. Genes Dev 2006; 20(1):1-15; PMID:16391229; http://dx.doi.org/10.1101/gad.1376506
- Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev 2007; 87(1):99-163; PMID:17237344; http://dx.doi.org/10.1152/physrev.00013.2006
- Halestrap AP. Calcium, mitochondria and reperfusion injury: a pore way to die. Biochem Soc Trans 2006; 34(Pt 2):232-7; PMID:16545083
- Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Pan H, Kessler H, Pancoska P, Moll UM, et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 2005; 434(7033):658-62; PMID:15800627; http://dx.doi.org/10.1038/nature03434
- Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, Inohara H, Kubo T, Tsujimoto Y. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 2005; 434(7033):652-8; PMID:15800626; http://dx.doi.org/10.1038/nature03317
- Schinzel AC, Takeuchi O, Huang Z, Fisher JK, Zhou Z, Rubens J, Hetz C, Danial NN, Moskowitz MA, Korsmeyer SJ. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc Natl Acad Sci U S A 2005; 102(34):12005-10; PMID:16103352; http://dx.doi.org/10.1073/pnas.0505294102
- Basso E, Fante L, Fowlkes J, Petronilli V, Forte MA, Bernardi P. Properties of the permeability transition pore in mitochondria devoid of Cyclophilin D. J Biol Chem 2005; 280(19):18558-61; PMID:15792954; http://dx.doi.org/10.1074/jbc.C500089200
- Tu HC, Ren D, Wang GX, Chen DY, Westergard TD, Kim H, Sasagawa S, Hsieh JJ, Cheng EH. The p53-cathepsin axis cooperates with ROS to activate programmed necrotic death upon DNA damage. Proc Natl Acad Sci U S A 2009; 106(4):1093-8; PMID:19144918; http://dx.doi.org/10.1073/pnas.0808173106
- Nijboer CH, Heijnen CJ, van der Kooij MA, Zijlstra J, van Velthoven CT, Culmsee C, van Bel F, Hagberg H, Kavelaars A. Targeting the p53 pathway to protect the neonatal ischemic brain. Ann Neurol 2011; 70(2):255-64; PMID:21674585; http://dx.doi.org/10.1002/ana.22413
- Du H, Guo L, Fang F, Chen D, Sosunov AA, McKhann GM, Yan Y, Wang C, Zhang H, Molkentin JD. et al. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer's disease. Nat Med 2008; 14(10):1097-105; PMID:18806802; http://dx.doi.org/10.1038/nm.1868
- Zhao LP, Ji C, Lu PH, Li C, Xu B, Gao H. Oxygen glucose deprivation (OGD)re-oxygenation-induced in vitro neuronal cell death involves mitochondrial cyclophilin-DP53 signaling axis. Neurochem Res 2013; 38(4):705-13; PMID:23322110; http://dx.doi.org/10.1007/s11064-013-0968-5
- Zhen YF, Wang GD, Zhu LQ, Tan SP, Zhang FY, Zhou XZ, Wang XD. P53 dependent mitochondrial permeability transition pore opening is required for dexamethasone-induced death of osteoblasts. J Cell Physiol 2014; 229(10):1475-83; PMID:24615518
- Chen B, Xu M, Zhang H, Wang JX, Zheng P, Gong L, Wu GJ, Dai T. Cisplatin-induced non-apoptotic death of pancreatic cancer cells requires mitochondrial cyclophilin-D-p53 signaling. Biochem Biophys Res Commun 2013; 437(4):526-31; PMID:23845906; http://dx.doi.org/10.1016/j.bbrc.2013.06.103
- Lu JH, Shi ZF, Xu H. The mitochondrial cyclophilin Dp53 complexation mediates doxorubicin-induced non-apoptotic death of A549 lung cancer cells. Mol Cell Biochem 2014; 389(1-2):17-24; PMID:24343341; http://dx.doi.org/10.1007/s11010-013-1922-1
- Montero J, Dutta C, van Bodegom D, Weinstock D, Letai A. p53 regulates a non-apoptotic death induced by ROS. Cell Death Differ 2013; 20(11):1465-74; PMID:23703322; http://dx.doi.org/10.1038/cdd.2013.52
- Endo H, Saito A, Chan PH. Mitochondrial translocation of p53 underlies the selective death of hippocampal CA1 neurons after global cerebral ischaemia. Biochem Soc Trans 2006; 34(Pt 6):1283-6; PMID:17073802
- Wang W, Cheng X, Lu J, Wei J, Fu G, Zhu F, Jia C, Zhou L, Xie H, Zheng S. Mitofusin-2 is a novel direct target of p53. Biochem Biophys Res Commun 2010; 400(4):587-92; PMID:20804729; http://dx.doi.org/10.1016/j.bbrc.2010.08.108
- Li J, Donath S, Li Y, Qin D, Prabhakar BS, Li P. miR-30 regulates mitochondrial fission through targeting p53 and the dynamin-related protein-1 pathway. PLoS Genet 2010; 6(1):e1000795; PMID:20062521; http://dx.doi.org/10.1371/journal.pgen.1000795
- Bae BI, Xu H, Igarashi S, Fujimuro M, Agrawal N, Taya Y, Hayward SD, Moran TH, Montell C, Ross CA. et al. p53 mediates cellular dysfunction and behavioral abnormalities in Huntington's disease. Neuron 2005; 47(1):29-41; PMID:15996546;http://dx.doi.org/10.1016/j.neuron.2005.06.005
- Trettel F, Rigamonti D, Hilditch-Maguire P, Wheeler VC, Sharp AH, Persichetti F, Cattaneo E, MacDonald ME. Dominant phenotypes produced by the HD mutation in STHdh(Q111) striatal cells. Hum Mol Genet 2000; 9(19):2799-809; PMID:11092756; http://dx.doi.org/10.1093/hmg/9.19.2799
- Guo X, Disatnik MH, Monbureau M, Shamloo M, Mochly-Rosen D, Qi X. Inhibition of mitochondrial fragmentation diminishes Huntington's disease-associated neurodegeneration. J Clin Invest 2013; 123(12):5371-88; PMID:24231356; http://dx.doi.org/10.1172/JCI70911
- Leu JI, George DL. Hepatic IGFBP1 is a prosurvival factor that binds to BAK, protects the liver from apoptosis, and antagonizes the proapoptotic actions of p53 at mitochondria. Genes Dev 2007; 21(23):3095-109; PMID:18056423; http://dx.doi.org/10.1101/gad.1567107