
ABSTRACT
Nicotinamide phosphoribosyltransferase (NAMPT) is crucial for nicotinamide adenine dinucleotide (NAD+) biosynthesis in mammalian cells. NAMPT inhibitors represent multifunctional anticancer agents that act on NAD+ metabolism to shut down glycolysis, nucleotide biosynthesis, and ATP generation and act indirectly as PARP and sirtuin inhibitors. The selectivity of NAMPT inhibitors preys on the increased metabolic requirements to replenish NAD+ in cancer cells. Although initial clinical studies with NAMPT inhibitors did not achieve single-agent therapeutic levels before dose-limiting toxicities were reached, a new understanding of alternative rescue pathways and a biomarker that can be used to select patients provides new opportunities to widen the therapeutic window and achieve efficacious doses in the clinic. Recent work has also illustrated the potential for drug combination strategies to further enhance the therapeutic opportunities. This review summarizes recent discoveries in NAD+/NAMPT inhibitor biology in the context of exploiting this new knowledge to optimize the clinical outcomes for this promising new class of agents.
KEYWORDS:
Abbreviations:
| 5-FU, | = | 5-Fluorouracil; |
| AIF, | = | apoptosis inducing factor; |
| ARTs, | = | mono ADP-ribose transferases; |
| ATP, | = | adenosine triphosphate; |
| AML, | = | acute myeloid leukemia; |
| AMP, | = | adenosine monophosphate; |
| BER, | = | base excision repair; |
| cADP, | = | cyclic adenosine diphosphate; |
| CLL, | = | chronic lymphocytic leukemia; |
| Cx43, | = | connexin 43; |
| dTTP, | = | deoxythymidine triphosphate; |
| dUTP, | = | deoxyuridine triphosphate; |
| FFPE, | = | formalin fixed paraffin embedded; |
| GI, | = | gastrointestinal; |
| HDAC, | = | histone deacetylase; |
| LDHA, | = | lactate dehydrogenase A; |
| NA, | = | nicotinic acid; |
| NAAD, | = | nicotinic acid adenine dinucleotide; |
| NAD+, | = | nicotinamide adenine dinucleotide (oxidized); |
| NADH, | = | nicotinamide adenine dinucleotide (reduced); |
| NADP+, | = | nicotinamide adenine dinucleotide phosphos-phorylated (oxidized); |
| NADPH, | = | nicotinamide adenine dinucleotide phosphosphorylated (reduced); |
| NADS, | = | NAD synthetase; |
| NAMN, | = | nicotinic acid mononucleotide; |
| NAMPT, | = | nicotinamide phosphoribosyltransferase; |
| NAMPTi, | = | NAMPT inhibitor; |
| NAPRT1, | = | nicotinic acid phosphoribosyl transferase 1; |
| NM, | = | nicotinamide; |
| NMN, | = | nicotinamide mononucleotide; |
| NMNAT, | = | nicotinamide mononucleotide adenylyltransferase; |
| NQO1, | = | NAD(P)H dehydrogenase [quinone] 1; |
| NR, | = | nicotinamide riboside; |
| NRK, | = | nicotinamide riboside kinase; |
| NSCLC, | = | non-small cell lung carcinoma |
| PAR, | = | poly-ADP–ribose; |
| PARP, | = | poly-ADP–ribose polymerase; |
| QA, | = | quinolinic acid; |
| QAPRT, | = | quinolinic acid phosphoribosyltransferase; |
| ROS, | = | reactive oxygen species; |
| SIRT, | = | sirtuin; |
| TCA, | = | tricarboxylic acid cycle; |
| TRP, | = | tryptophan |
Introduction
Interfering with nicotinamide adenine dinucleotide (NAD+) biosynthesis as a therapeutic strategy for cancer holds great promise. The key NAD+ biosynthetic enzyme involved in NAD+ generation is nicotinamide phosphoribosyltransferase (NAMPT) and several inhibitors are currently in various phases of development as anticancer agents. Within the last decade, 2 different classes of NAMPT inhibitors (NAMPTis) have been tested in clinical trials as single-agent therapies but did not show efficacy. Since that time, a great deal more has been learned about the regulation of NAD+ in cancer cells, in particular the contribution of parallel pathways and regulatory enzymes. This new information will have a profound effect on the types of therapeutic opportunities that exist and the design of future clinical trials for NAMPTis.
Role of metabolism in cancer
Cellular metabolic adaptation is a key requirement for malignant cell transformation. The increased proliferation of transformed cells and the requirement for continual DNA, protein, and lipid biosynthesis generate high demands for the energy (ATP) required to drive these processes. Some cancer-specific metabolic adaptations include increased rates of aerobic glycolysis and oxidative phosphorylation and increased uptake of glucose and glutamine, with the goal of fulfilling these needs.Citation1,2 NAD+/NADH and their phosphorylated forms (NADP+/NADPH) are key to this adaptation process as they act as enzymatic co-factors that are rapidly oxidized and reduced to generate ATP and counteract intracellular oxidative stress. In the last decade NAD+ has also been implicated as the substrate for important cancer-related enzymes such as ADP-ribose transferases, including poly-ADP-ribose polymerases (PARPs), sirtuins, and cyclic ADP (cADP) ribose synthases. These enzymes are important in DNA repair, G-protein coupled receptor signaling, calcium homeostasis, transcriptional regulation, and ultimately cancer cell survival.Citation2,3 Unlike the redox reactions, which reversibly oxidize or reduce pyridine nucleotides as co-factors, enzymes that use NAD+ as a substrate continuously consume NAD+ and therefore play a crucial role in cancer metabolism as regulators of NAD+/NADH availability. Nicotinamide (NM) released during NAD+- dependent reactions must be recycled to maintain tissue NAD+ levels; this occurs mainly through the NAMPT pathway.
Mammalian NAD+ synthesis pathways
In mammalian cells, NAD+ is either synthesized de novo from the amino acid tryptophan or through one of 3 salvage pathways utilizing NM, nicotinic acid (NA), or nicotinamide riboside (NR) as precursors (reviewed in references 1, 2; ). The de novo pathway converts dietary tryptophan through a series of enzymatic steps in the kynurenine pathway leading to quinolinic acid (QA), which is then converted to nicotinic acid mononucleotide (NAMN) via quinolinic acid phosphoribosyltransferase (QAPRT). This pathway plays a role in NAD+ generation primarily in the liver, immune tissues, and the brain under inflammatory or stress conditions (reviewed in references 3, 4).
Figure 1. Pathways involved in NAD+ biosynthesis and catabolism. Metabolites: NA, nicotinic acid or niacin; TRP, tryptophan; QA, quinolinic acid; NAMN, NA mononucleotide; NAAD, nicotinic acid adenine dinucleotide; NAD+, nicotinamide adenine dinucleotide (oxidized); NM, nicotinamide; NMN, NM mononucleotide; NR, nicotinamide riboside. Metabolic enzymes are: NAPRT1, nicotinic acid phosphoribosyltransferase; NMNAT1,2,3, nicotinamide nucleotide adenylyltransferases; NADS, NAD+ synthetase; PARPs, poly ADP-ribose polymerases; SIRTs, sirtuins; CD38, cluster of differentiation 38 or cyclic ADP-ribose hydrolase; NAMPT, nicotinamide phosphoribosyl transferase; NRK1,2, nicotinamide riboside kinases; Cx43, connexin43; CD73, cluster of differentiation 73 or ecto-5′-nucleotidase.
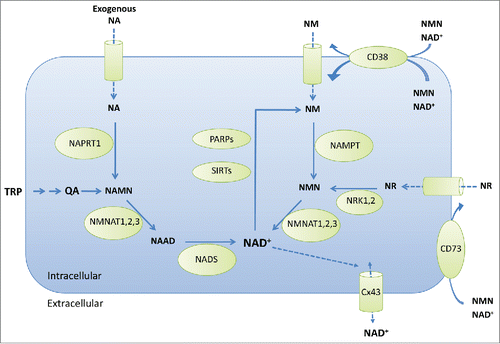
Among the 3 mammalian salvage pathways, the primary pathway to NAD+ synthesis is a 2-step conversion, first from NM to nicotinamide mononucleotide (NMN) and then from NMN to NAD+. The first step is rate limiting and involves the cytosolic enzyme NAMPT.Citation5 The second step involves a family of 3 enzymes, nicotinamide mononucleotide adenylyltransferases (NMNAT) 1, 2, and 3, which are localized to the nucleus, Golgi, and mitochondria respectively.Citation6 NAMPT is infrequently mutated in normal and cancer cellsCitation7 and is upregulated in response to lymphocyte activationCitation8 and cellular stresses such as nutrient deprivation,Citation9 consistent with its essential role in cellular metabolism and metabolic adaptation. Overall, in mice the liver and kidney contribute the highest levels of NAD+ synthesis through the NAMPT salvage pathway.Citation10 In the second salvage pathway the precursor nicotinamide riboside (NR) is phosphorylated by NR kinases 1 and 2 to give nicotinamide mononucleotide (NMN), which is then converted to NAD+ as above.Citation11 The third salvage route to NAD+ synthesis is via the Preiss-Handler pathway that utilizes NA and requires the expression of nicotinic acid phosphoribosyltransferase 1 (NAPRT1) for the first enzymatic step to convert NA to nicotinic acid mononucleotide (NAMN).Citation12 NAMN is converted through the NMNATs to nicotinic acid adenine dinucleotide (NAAD), and then to NAD+ by NAD+ synthetase. The NMNAT enzymes are widely distributed and considered non-rate limiting for conversion of either NAMN or NMN.Citation10 NAD+ synthetase may exhibit a more narrow tissue expression and activity range, being virtually undetectable in the lung and skeletal muscle but highly expressed in the liver and kidney.Citation10
Interestingly, NAMPT also exists in an extracellular form known as the adipocytokine visfatin present in the circulation;Citation13 however, the contribution of visfatin to extracellular NAD+ metabolism is still controversial as studies have reported conflicting levels of the enzymatic product NMN in plasmaCitation14,15 and some results suggest that ATP concentrations in the extracellular environment are not sufficient to permit enzymatic activity.Citation15 The additional biologic effects of visfatin extend beyond the scope of this review but have been reviewed elsewhere.Citation13
NAD+ consuming enzymes
NAD+ consuming enzymes, which are highly active in many cancers, are key regulators of intracellular NAD+ availability. Inhibitors of NAD+ synthesis have a direct impact on the functionality of these proteins. There are 4 major classes of NAD+ consuming enzymes: the intracellular PARPs and sirtuins, and the extracellular mono ADP-ribose transferases (ARTs) and cADP ADP-ribose synthetases. Relatively little is known about the ARTs in the context of their impact on NAD+ metabolism and cancer and they are not discussed extensively here.
Intracellular enzymes
PARPs
PARPs are important regulators of cell stress and responses to DNA damage (reviewed in references 16 and 17). There are 17 known PARPs, but PARP-1, the best studied and most enzymatically active in vitro, is thought to be the major contributor to the DNA damage response. PARP-1 utilizes NAD+ as the substrate for auto poly-ADP-ribosylation (PARylation) and activation, followed by the addition of hundreds of poly-ADP-ribose (PAR) subunits in chains to target proteins such as histones, transcription factors, and DNA repair proteins. PARPs are highly responsive proteins; their enzymatic activity can increase up to 500-fold within seconds after detecting DNA damage and they can consume up to 80–90% of the intracellular NAD+ stores.Citation17,18 PARPs are able to recognize both single- and double-strand DNA breaks and recruit the appropriate protein complexes to effect the 4 major DNA repair pathways (nucleotide excision repair, base excision repair [BER], homologous recombination, and non-homologous end joining).Citation19 PARP is overexpressed in several cancers, including those with defective DNA repair pathways such as BRCA1/2 mutant tumors.Citation16
Sirtuins
Sirtuins (SIRTs) represent a family of 7 different conserved NAD+ dependent deacetylases that mediate their effects by deacetylating transcription factors and histones, causing changes in the expression of genes involved in a wide variety of cellular processes including the response to nutritional and energy status, DNA damage response, oxidative stress, lipid metabolism, and cell death. Sirtuins have been implicated positively and negatively as regulators of tumorigenesis depending on the individual sirtuin involved (reviewed in references 20, 21). For example, SIRT1 is upregulated in many cancer types and is thought to promote tumor development and progression by deacetylating and inactivating the tumor suppressors p53 and FOXO, both of which regulate proapoptotic gene expression.Citation20
Extracellular enzymes
cADP ribose synthetases
More recently, 2 ectoenzymes have been implicated in the regulation of NAD+ metabolism: CD38 and CD73 (or 5´-nucleotidase). CD38 is mainly expressed in hematopoietic cells including T and B lymphocytes, monocytes, and natural killer (NK) cells and is also expressed at lower levels in non-immune tissue (reviewed in 22, 23). Upregulated CD38 expression serves as a marker for advanced stage and poor prognosis in chronic lymphocytic leukemia (CLL) and multiple myeloma.Citation24,25 Varying, but elevated, levels of CD38 expression have also been found in pancreatic cancer patient samples compared to normal pancreatic cells.Citation26 CD38 acts as a NADase, hydrolyzing NAD+ or NMN to NM and ADP-ribose or cADP-ribose, which serve as important modulators of intracellular Ca2+ signaling.Citation27 Although it is mostly characterized as a type II membrane protein whose catalytic domain is extracellular, a portion of CD38 is expressed in the opposite orientation such that the catalytic domain is on the cytosolic side. CD38 is also internalized into endocytic vesicles,Citation23 permitting conversion of intracellular NAD+ and NMN to NM. CD38 knockout mice have elevated levels of NAD+ in some tissues, pointing to the constitutive nature of this NAD+ consuming enzyme.Citation26
The link between CD73 and tumorigenesis is variable and may also be context dependent. The activity of the enzyme is high in malignancies of epithelial origin, particularly glioblastoma.Citation28 Although the best-known function of CD73 is the conversion of AMP to adenosine, recent work demonstrated that purified human CD73 converts NAD+ and NMN (NMN being the preferred substrate) to NR in vitro.Citation29 The in vivo contribution of this enzyme to conversion of NAD+ and NMN in the extracellular space has yet to be demonstrated.
NAD+ metabolites
Little is known about physiologic levels of various NAD+ metabolites in circulation or in tissues. In the plasma, NM is found at low μM levels whereas NA is undetectable and highly labile.Citation30 Circulating levels of other NAD+ precursors such as NR and NMN are largely unknown and have been difficult to quantify since they are often unstable or present at very low levels. NAD+ can be imported and exported from cells through Connexin 43 (Cx43) hemichannels to allow access to the ectoenzymes that can metabolize it, potentially leading to paracrine effects on nearby cells.Citation31 However, understanding NAD+ metabolism in the interstitial space will undoubtedly be challenging. The mechanism by which NAD+ biosynthesis is regulated is predicted to be tissue specific, based on access to extracellular metabolites and the expression patterns of the metabolic enzymes involved.
In cells, although NAMPT is mainly localized to the nucleocytosolic compartment, mitochondria house the complexes of enzymes that require NAD+/NADH in the TCA cycle and electron transport chain to generate ATP, and accordingly contain a large proportion (up to 70%) of the intracellular NAD+ stores.Citation32 Mitochondrial NAD+ levels are the determining factor for cell survival and are carefully preserved and regulated by sirtuins.Citation33 The NAD metabolite NMN, and possibly NAD+, can be imported from the cytosol and converted by mitochondrial NMNAT3 to replenish intra-mitochondrial NAD+ pools.Citation34
NAMPT as a cancer target
NAMPT has several well-established links to cancer including its differential expression in tumor tissue and its correlation with cancer therapy resistance and poor outcome in cancer patients. Colorectal,Citation35 ovarian,Citation36 and prostateCitation37 carcinomas, astrocytomasCitation38 and melanomaCitation39 tumors overexpress NAMPT relative to their benign counterparts. Higher levels of NAMPT expression correlate with more aggressive lymphomasCitation40 and high-grade astrocytomas,Citation38 and with poor outcome in gastricCitation41 and endometrial carcinomas. Citation42 There is evidence that conditional c-MYC overexpression induces upregulation of NAMPT gene expression in colorectal cancers.Citation43 NAMPT is overexpressed in bortezomib-resistant multiple myeloma cellsCitation44 and is associated with resistance to etoposide in vitro.Citation9 Inhibition of NAMPT activity by small molecule inhibitors or gene knockdown has demonstrated antitumor activities both in vitro and in multiple in vivo models of human cancer including prostate cancer, pancreatic cancer, ovarian cancer, fibrosarcoma, small cell and non-small cell lung cancer (NSCLC), colon cancer, and myeloid leukemia.Citation37,45-52 Tumor cell sensitivity to NAMPTis are inversely proportional to the level of NAMPT expression.Citation40,46,48,53
Cellular effects of NAMPT inhibition
Metabolic effects
The first-generation NAMPTis, APO866/FK866 (Apoxis/Topotarget) and CHS-828 (Leo Pharma), later renamed GMX1778 [Gemin X/Cephalon/Teva]), are inhibitors that have been tested in clinical trials and are the bestcharacterized in the literature. GMX1777 (formerly EB1627) is a soluble prodrug of GMX1778 that was used for clinical trials.Citation54 More recently developed inhibitors include GNE-617,Citation55 GNE-618,Citation56 and several others that have been published in the scientific and patent literature (extensively reviewed in references 57, 58). The APO866, GMX1778, and GNE-617/8 compounds represent distinct chemical classes and each is a competitive inhibitor with low nanomolar potency.Citation45,46,48,52,59 Common to highly potent NAMPTis is their ability to bind within the canal-shaped NM substrate binding site and become phosphoribosylated by NAMPT, resulting in a tighter interaction and more potent inhibitory activity (reviewed in reference 58). This has been demonstrated for GMX1778 and GNE-618.Citation46,60 The effects of NAMPT inhibition on multiple metabolic pathways are profound. Metabolomic analysis of cells treated with NAMPTis in vitro reveals a rapid decrease in intracellular NAD+ and mild inhibition of glycolysis and nucleotide metabolism within the first 6–10 hours of treatment.Citation46,48 This is followed by a range of secondary metabolic effects including more profound inhibition of glycolysis, decreased carbon flux through the TCA cycle and subsequent ATP synthesis, inhibition of aspartate and alanine metabolism, inhibition of purine and pyrimidine metabolism, and decreased guanylate nucleotide synthesis at 24 h.Citation61,62 Cell death follows a >90% reduction in ATP and begins 36–48 h after treatment initiation.Citation46,50,61,63 Similar pathway perturbations to glycolysis, nucleotide, and phospholipid biosynthesis were seen in vivo in tumors from mice treated with APO866.Citation64
In A549 cells faced with sub-lethal doses of NAMPTi, NAD+ levels decline with the same kinetics as with higher doses but then recover between 24–48 h post treatment from an almost complete loss of NAD+, suggesting an active adaptation to fluctuating NAD+ levels.Citation50 There is clearly a threshold level and/or duration of NAD+ depletion after which treated cells cannot survive. Supporting this hypothesis is the fact that dose-response curves of NAMPTi treatment in cell lines in vitro are very steep, suggesting that a defined threshold of NAD+ depletion must be attained for cytotoxic effectCitation46 and, furthermore, that sustained exposure of to NAMPTi for more than 48 h, and subsequent NAD+ depletion, is required for maximal cytotoxicity.Citation49 This has been demonstrated in vivo, where doses of GNE-617 sufficient to reduce NAD+ levels in HT1080 tumors to 80% of those in vehicle-treated animals were not sufficient to effect an antitumor response. However, consistent with in vitro data, doses sufficient to achieve a >90% reduction in NAD+ resulted in cytotoxicity and a robust antitumor response.Citation50,52 Interestingly, Pitteli et al. demonstrated that APO866 inhibits cytosolic and PARP-dependent NAD+ levels in cells while leaving the mitochondrial NAD+ pool intact, suggesting this may be the last location in which NAD+ is depleted.Citation65 Understanding the metabolic determinants of this cell survival threshold in cancer versus normal cells will be important for establishing a therapeutic index and for the study of NAMPTi resistance.
Although some studies have characterized the mode of NAMPT-induced cell death as either apoptosis or autophagy,Citation45,66 Del Nagro et al.Citation63 found that the death inducing pathway depends on the rate of ATP depletion. Cells in which there was a delay between NAD+ and ATP depletion died of apoptosis or autophagy, whereas cells in which ATP depletion occurred immediately after NAD+ depletion died of oncosis, a pathway characterized by cellular swelling and the appearance of blisters in the plasma membrane.
Effects on PARPs and sirtuins
In view of the fact that NAD+ is a crucial substrate for PARPs and sirtuins, it is not surprising that changes in NAD+ levels can affect the activity of these enzymes. NAMPTis, such as GMX1777/8, can effectively inhibit PARP activity by depleting cellular NAD+ levels.Citation50,67,68 In theory, this could allow cells that have recovered from incomplete NAD+ depletion to accrue unrepaired DNA lesions that will eventually lead to cell death, and there is evidence for this as cells treated with sublethal doses of NAMPTis in the presence of DNA damaging agents exhibit accumulation of double-strand DNA breaks (marked by γ-H2AX) suggesting a potential inhibitory effect on DNA repair.Citation50,68 In this respect, NAMPTis can act as PARP inhibitors; indeed, they effectively combine with some of the agents that also synergize with PARP inhibitors (e.g., radiation therapy or temozolomide).Citation67,69-71
As is the case with PARP, NAMPTis can also indirectly inhibit SIRT activity by depleting the NAD+ substrate on which they depend for activity.Citation37,72 In keeping with this, NAMPT inhibition causes hyperacetylation of α-tubulin and histone H3 in lung cancer cells.Citation63,73 In prostate cancer cells, APO866 inhibits global protein acetylation (through inhibition of SIRT1 and SIRT3 activity) and subsequently the fatty acid and phospholipid biosynthesis required for their survival.Citation74
The altered metabolic activity and higher levels of reactive oxygen species (ROS) in cancer cells render them more dependent on sensing and responding to oxidative damage.Citation75 Thus, in addition to depleting NAD+, NAMPTis may indirectly have anticancer activity through inhibition of enzymes such as PARPs and sirtuins that mediate the response to ROS and the repair of ROS-mediated DNA damage.
Resistance mechanisms
Some information has been accumulated about the potential mechanisms of resistance to NAMPTis. Several NAMPT mutations have been identified in cell lines treated with inhibitors that permit sufficient functional NAMPT activity for cell survival yet preclude inhibitor binding through steric hindrance of the nicotinamide/drug binding site.Citation46,53,76,77 The detected mutations in residues 93, 191, 217, and 388 occur in or near the nicotinamide binding site. Interestingly, NAMPTis of different classes have differing sensitivities to cells overexpressing these mutants.Citation76 An additional mutation at residue 165 precludes binding of the phosphoribosylated GNE-618 compound to the enzyme at an allosteric site; however, this mutation preferentially affects some compound classes over others.Citation76 None of these mutations were identified in a screen of more than 200 normal and tumor tissues, suggesting the lack of pre-existing resistance mutations in patient populations before treatment.Citation7 Given the plasticity of cancer cells, however, adaptive responses to declining intracellular NAD+ levels can be expected; therefore, NAMPTis that can avoid these potential resistance mechanisms will have great potential, especially for the treatment of patients in the relapsed setting.
Clinical experience with NAMPT inhibitors
More than 100 patients were treated in the first clinical trials with CHS-828 (later known as GMX1778 after it was determined to be a selective NAMPTi),Citation46 GMX1777, and APO866.Citation7-82 CHS-828 was administered orally, with the most frequently observed toxicities being thrombocytopenia and gastrointestinal (GI) effects such as diarrhea, vomiting, and esophagitis.Citation79,81 The considerable inter- and intrapatient variability in the pharmacokinetics of the orally administered agent made it clear that another formulation or dosing route would be needed,Citation81 therefore a pro-drug of CHS-828/GMX1778, EB1627 (later renamed GMX1777), was generated for intravenous administration.Citation54 GMX1777 and APO866 were administered by continuous intravenous infusion for 24 h or 96 h respectively. Dose-limiting toxicities for these agents also included thrombocytopenia and some GI effects. Additional adverse effects for GMX1777 and APO866 seen in some patients included skin rash and lymphopenia.Citation78,80 Oral administration of GMX1778 tended to generate more frequent adverse GI effects than the 2 intravenously administered agents APO866 or GMX1777.Citation82 No objective responses were seen in the CHS-828 or APO866 trials with advanced solid tumors or in the GMX1777 trial with advanced malignancies, although some patients exhibited stable disease.Citation78-80 One possible reason for the lack of efficacy is that insufficiently high doses of NAMPT inhibitor could be achieved for clinical efficacy when administered as a single agent. Another possibility is that the dosing schedule was not optimized to attain the threshold duration and degree of NAD+ depletion required to cause cytotoxicity in tumor cells.
Potential combination strategies for the clinical use of NAMPT inhibitors
The growing list of agents that enhance the effectiveness of NAMPTis when administered in combination is summarized in . As described below, promising combination strategies are likely to involve biomarkers for patient selection in addition to co-administration of a second agent.
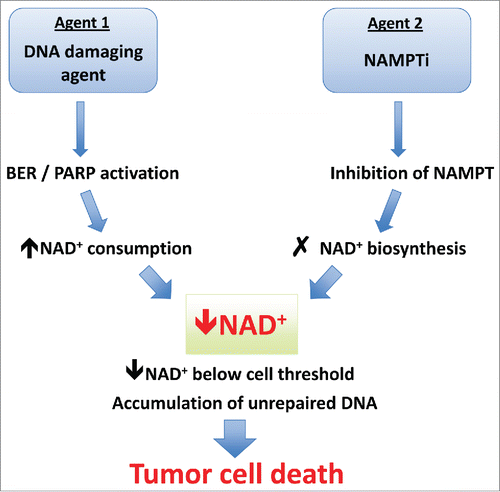
Figure 2. Mechanism of synergy between DNA damaging agents and NAMPT inhibitors. NAMPTi synergize with several DNA damaging agents, all of which have been shown to induce base excision repair (BER) or single-stranded DNA damage resulting in PARP activation. The increased consumption of NAD+ caused by PARP activation combined with inhibition of NAD+ regeneration leads to catastrophic NAD+ depletion and cell death. In cells that do not reach the threshold level/duration of NAD+ depletion, accumulation of unrepaired DNA may later lead to cell cycle arrest and tumor cell death. BER, base excision repair; NAD+, nicotinamide adenine dinucleotide (oxidized); NAMPT, nicotinamide phosphoribosyl transferase; NAMPTi, NAMPT inhibitor; PARP, poly ADP-ribose polymerase.
Niacin
Niacin (NA or vitamin B3) is commonly used as a dietary supplement and to treat pellagra, a disease caused by vitamin B3 deficiency. Niacin is also used at higher doses in the cardiovascular setting to treat dyslipidemia or high cholesterol.Citation83 Early studies with NAMPTis demonstrated that cellular NAD+ depletion and cytotoxicity could be rescued through the Preiss-Handler pathway by exogenous addition of NA.Citation45 Rescue through this pathway was determined to be solely dependent on NAPRT1 expression.Citation46,51,84 Since NAPRT1 is expressed in the majority of tissues in mammals, one potential strategy for safely enabling clinically effective doses of NAMPTi is to combine them with niacin administration.Citation85 Of course, in order to effectively implement such a strategy therapeutically it would be necessary to bypass niacin rescue of NAMPTi in the tumor. Studies in mice demonstrate that co-administration of niacin with lethal doses of NAMPTi rescued mortality, in addition to toxicities such as thrombocytopenia and lymphopenia.Citation46,51 Niacin co-administration also minimized histologic signs of toxicity to testis, spleen, and lymphoid tissue, and reversed the toxicity to the less-affected liver, kidney and gastrointestinal tissues, thus ameliorating many of the toxicities that were observed in human clinical trials.Citation51 In vitro, NA, kynurenine, and NAD+ were able to fully rescue APO866-induced cell death of human, but not murine, lymphocytes, highlighting the potential species-specific nature of NAD+ metabolic pathways in different tissues or the higher affinity of this NAMPT inhibitor for murine NAMPT compared to human.Citation86 Recent rat studies demonstrate that retinal toxicity is induced after 4 days of treatment with GNE-617/618 or GMX1778. This toxicity is not species specific, is on-target, and is not rescued by niacin co-administration.Citation87 Additionally, some cardiotoxicity that has been observed with a similar dosing schedule was partially rescued by NA administration.Citation58 It remains to be determined whether a sustained 4-day treatment regimen is necessary for antitumor activity given that some studies have demonstrated antitumor activity after a single 24-h continuous infusion.Citation49 Further study will be required to determine whether adjusting the niacin dose and the duration of NAMPTi treatment will be able to overcome these toxicities. Clearly, NAMPTis that do not penetrate the blood-brain or blood-retinal barriers may have some additional safety-related advantages.
As noted above, in order for the niacin rescue strategy to be optimally effective at targeting tumor tissue while protecting non-cancerous host tissues, the tumors must be immune to niacin rescue and therefore NAPRT1 negative. In fact, a variety of tumor types do not express NAPRT1, thus providing a strategy for selection of patients whose tumors are not rescued by co-administration of niacin with therapeutic NAMPTi doses.Citation46,84 The feasibility of this strategy was demonstrated in murine models using NAPRT1-negative xenografts. In two studies, HT1080 and A2780 xenografts were not rescued when modest niacin doses (below those used to treat hyperlipidemia) were co-administered with NAMPTis at doses above their maximum tolerated doses.Citation46,51 In these studies, greater efficacy was achieved by allowing a higher dose of NAMPTi to be tolerated. In other examples, when higher or prolonged niacin doses were co-administered with lower NAMPTi doses (at their single-agent maximum tolerated dose) the antitumor efficacy for NAPRT1-negative tumors was either completely or partially inhibited, presumably because of elevated levels of circulating NAD+ metabolites (including NM) released from the liver of treated animals.Citation52 In general, NAPRT1-positive xenograft tumor growth is rescued by niacin co-administration, indicating the essential requirement for selection of patients with NAPRT1-negative tumors for this strategy.Citation46,48,84 Interestingly, when niacin administration was delayed to 24 h after a short (24 h) treatment with NAMPTi, even the NAPRT1-positive tumors showed a substantial antitumor response with the niacin co-administration strategy.Citation49 This further indicates that the timing and duration of niacin and NAMPTi treatment may contribute to the potential success of this combination strategy in the clinic.
The frequency of cell lines from multiple tumor origins lacking NAPRT1 expression is wide-ranging and substantial. Screens of panels of cell lines suggest that neurologic tumors (glioblastoma and neuroblastoma) and various sarcomas have the highest incidence, with more than 40% NAPRT1-negative cells based on NAPRT1 protein expression and NA rescue status.Citation46,84 Furthermore, NAPRT1 mRNA levels are directly proportional to lack of NA rescueCitation46,84 and indicate that more than 15% of liver, lymphoma, ovarian, multiple myeloma, and NSCLC cell lines have very low NAPRT1 gene expression.Citation84 Similar frequencies were confirmed by histologic examination of formalin-fixed paraffin-embedded (FFPE) samples from patients with small cell lung cancer, Hodgkin's lymphoma, and non-Hodgkin's lymphoma.Citation84 Lack of NAPRT1 gene expression was not derived from gene deletion and only infrequently caused by loss of heterozygosity, but instead was determined to be mainly due to promoter hypermethylation.Citation84 This led to the development of a potential quantitative methylation-specific PCR assay for the detection of NAPRT1-negative tumors from FFPE samples. The high frequency of this potential biomarker across multiple indications provides ample opportunities for patient selection, as NAMPTis will almost certainly not be used clinically in the absence of niacin co-administration.
DNA damaging agents
As PARylation has been suggested to be the most important factor determining NAD+ catabolism in cells, the activation of PARP-1 in response to DNA damaging agents is probably one of the major causes of rapid NAD+ consumption.Citation18 Cells must respond to this consumption by resynthesizing NAD+ as quickly as possible through the NAMPT-mediated NM recycling pathway. If this salvage pathway is blocked by NAMPT inhibition, it seems logical that any agent that can activate PARPs to appreciable levels could be combined with NAMPTis to yield synergistic cytotoxic activity. Several examples of this are summarized in . and the mechanism is illustrated in . For example, GMX1777 and APO866 enhance sensitivity to radiotherapy in head and neck cancer and prostate cancer through NAMPTi-mediated NAD+ depletion and a radiation therapy-mediated increase in PARP activity.Citation67,70 This is in keeping with the proposed mechanism of synergy caused by a simultaneous increase in NAD+ consumption and inhibition of NAD+ regeneration. Through a similar mechanism, several DNA damage-inducing chemotherapies such as temozolomide,Citation71 5-fluorouracil (5-FU),Citation41 fludarabine,Citation88 pemetrexedCitation50 and β-lapachoneCitation68 are synergistic in combination with NAMPTis. Interestingly, the temozolomide combination works well in cells harboring 2 known mechanisms of temozolomide resistance, elevated expression of O6-methylguanine-DNA methyltransferase or a defect in mismatch repair.Citation71 Temolozomide and melphalan introduce DNA lesions with N-methylated or alkylated adducts, respectively, whereas 5-FU, fludarabine, and pemetrexed interfere with nucleotide metabolism, causing an imbalance in dUTP/dTTP pools and dUTP base misincorporation into newly synthesized DNA.Citation89,90 Both types of DNA adduct and base misincorporation are repaired by the base excision repair (BER) pathway that, if incomplete, results in double-strand DNA breaks and hyperactivation of PARP.Citation91 In tumor cells that express high levels of NADPH:quinone oxidoreductase (NQO1), such as pancreatic cancer cells, β-lapachone is converted to a highly reactive derivative that generates ROS and subsequent single-strand DNA breaks. This type of DNA damage is also repaired by hyperactivation of PARP1.Citation68 Some studies have suggested that DNA damage-induced PARP hyperactivation leads to cell death by accumulation of PAR polymers and translocation of apoptosis-inducing factor (AIF) from the mitochondria to the nucleus, leading to apoptosis.Citation92 This phenomenon does not occur with the combination of NAMPTis and agents such as pemetrexed or temozolomide because incomplete BER and PARP1 hyperactivation are rescued by the exogenous NAD+ precursor NMN independent of AIF translocation, supporting the conclusion that cell death during the response to NAMPT inhibition and DNA damaging agents results from NAD+ depletion.Citation50,68,91
Targeted inhibitors
NAMPT is a gene whose knockdown sensitizes cells to the PARP inhibitor olaparib, and APO866 synergizes with olaparib in several models of triple negative breast cancer.Citation93 Since APO866 alone highly depletes NAD+, the authors of this study were unable to demonstrate enhanced NAD+ depletion but suggested that the enhanced cytotoxic activity of the 2 agents may result from increased levels of double-strand breaks or enhanced inhibition of DNA repair leading to cytotoxicity. As NAD+ levels regulate a wide range of cellular processes, it might be expected that NAMPTis would also synergize with targeted agents not directly related to PARP or NAD+ metabolism (). Indeed, the NAMPT inhibitor APO866 synergizes with the histone deacetylase (HDAC) inhibitors valproic acid, butyrate, and vorinostat in primary AML and CLL cells.Citation94 Each agent contributes to the mechanism of synergy through Bax, a proapoptotic Bcl-2 family member. HDAC inhibition causes upregulation of Bax (possibly through SIRT1 inhibition and subsequent hyperacetylation of p53), while NAMPTi-mediated inhibition of SIRT1 relieves the cytosolic sequestration of Bax by Ku70, allowing it to migrate to mitochondria to effect cell death. Future prospects for NAMPTi drug combination strategies include dual inhibition of metabolic pathways, as in the lactate dehydrogenase A (LDHA) inhibitor/APO866 combination.Citation95 Finally, NAMPTi were also shown to synergize with TRAIL in primary CLL and T-cell leukemia cell lines,Citation96 and with the anti-CD20 monoclonal antibody rituxumabCitation97 and bortezomibCitation44 in multiple myeloma. The mechanism by which these agents cause synergy is unclear, although enhanced NAD+ depletion with the combination compared to each agent alone is a common feature.
Potential biomarkers for patient selection
In addition to lack of NAPRT1 expression (described above) as a likely essential biomarker for the use of NAMPTi, there are a few emerging biomarkers that may also predict sensitivity to this class of inhibitors. One is NAMPT itself, as several groups have demonstrated an inverse correlation between NAMPT expression levels and sensitivity to its inhibitors.Citation40,46,48,53 This presents something of a conundrum because an increase in NAMPT levels suggests that the cells have a greater dependence on NAD+ for survival, yet greater amounts of inhibitor are required to overcome the increased level of the NAMPT enzyme. Therefore, in cases where NAMPT is overexpressed, tumor cells require a concomitant induction of NAD+ consumption triggered by a second entity, such as a combination drug or activation of an NAD+ consuming enzyme, for maximum sensitivity to NAMPTis ().
Once candidate NAD+ consumer is the recently elucidated potential biomarker CD38. Consistent with the constitutive nature of CD38 enzymatic activity, overexpression of CD38 in pancreatic tumor cells increases their sensitivity to NAMPTis, presumably by enhancing NAD+ consumption.Citation47 In contrast, higher levels of CD38 correlate with lower sensitivity of CLL patient samples to NAMPT inhibition.Citation88 It is possible that cells that express high levels of CD38 for a period of time adjust by increasing NAMPT expression to support NAD+ synthesis, whereas in situations of transient overexpression the cells have not adapted to the increased NAD+ needs by upregulating NAMPT and are therefore more sensitive to NAMPTis. More work is necessary to determine whether CD38 will be a useful biomarker of cancer cell susceptibility to NAMPTis and whether it is context dependent. No studies have been published to date regarding the potential of CD73 as a marker of sensitivity or resistance.
Future directions
The promise of great therapeutic potential for agents that interfere with NAD+ metabolism has been strongly advanced by recent research in this area, which has increased exponentially within the last 5 years. However, the field of NAMPT biology is still in its infancy and there are many research avenues yet to be followed.
On the basis of what has been learned in recent years, NAMPTis have potential utility across multiple oncology indications and particularly in leukemia/lymphomas, multiple myeloma, glioblastoma, and lung and pancreatic cancers. Because NAMPTi have indirect inhibitory effects on PARP and sirtuins, cancer indications for which inhibitors of these enzymes are being explored should also be considered. It is clear from clinical experience with the first generation of NAMPTis that higher doses will be needed in order to achieve efficacy, and synthetic lethal drug combination strategies will therefore be essential. Co-administration with niacin appears to be an essential strategy designed to mitigate dose-limiting toxicities to normal tissues, thus enabling effective clinical NAMPTi levels to be achieved. Essential to the niacin co-administration strategy will be the selection of patients whose tumors are NAPRT1 negative. A better understanding of how NAD+ precursors such as niacin and their metabolites are distributed from central metabolizing centers (liver and kidney) to distal tissues will be important to predict how normal tissues and tumors will be affected by NAMPTis.
As adjuvants to niacin co-administration, additional drug combinations will also likely be part of the therapeutic strategy for NAMPTis. Certainly, chemotherapies that induce the BER DNA damage response have shown promise in preclinical models, but additional targeted therapies are beginning to emerge as potential combination approaches. As more is learned about the metabolic vulnerabilities of different tumor types, the best clinical strategies for NAMPTi/niacin/drug combinations will likely be ones that select patients whose tumors express a panel of susceptibility genes in addition to the lack of NAPRT1. For example, if DNA damage-inducing chemotherapies are part of the clinical strategy, one can imagine that the susceptibility genes may include DNA repair genes.
Finally, as new NAMPTis approach future clinical trials, selection of pharmacodynamic markers will be important to track compound activity in vivo and monitor vulnerable tissues for signs of toxicity.
Disclosure of potential conflicts of interest
AR and GCS were employees of Gemin X Pharmaceuticals Inc.; GCS was a shareholder in Gemin X Pharmaceuticals Inc.
Table 1. Potential therapeutic strategies for NAMPT inhibitors involving drug combinations.
Acknowledgments
This work was supported by grants from Genome Quebec and CIHR (MOP-6192) to GCS.
References
- Chiarugi A, Dolle C, Felici R, Ziegler M. The NAD metabolome–a key determinant of cancer cell biology. Nat Rev Cancer 2012; 12:741-52; PMID:23018234; http://dx.doi.org/10.1038/nrc3340
- Houtkooper RH, Canto C, Wanders RJ, Auwerx J. The secret life of NAD+: an old metabolite controlling new metabolic signaling pathways. Endocr Rev 2010; 31:194-223; PMID:20007326; http://dx.doi.org/10.1210/er.2009-0026
- Magni G, Orsomando G, Raffelli N, Ruggieri S. Enzymology of mammalian NAD metabolism in health and disease. Front Biosci 2008; 13:6135-54; PMID:18508649; http://dx.doi.org/10.2741/3143
- Dolle C, Skoge RH, Vanlinden MR, Ziegler M. NAD biosynthesis in humans–enzymes, metabolites and therapeutic aspects. Curr Top Med Chem 2013; 13:2907-17; PMID:24171775; http://dx.doi.org/10.2174/15680266113136660206
- Revollo JR, Grimm AA, Imai S. The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J Biol Chem 2004; 279:50754-63; PMID:15381699; http://dx.doi.org/10.1074/jbc.M408388200
- Berger F, Lau C, Dahlmann M, Ziegler M. Subcellular compartmentation and differential catalytic properties of the three human nicotinamide mononucleotide adenylyltransferase isoforms. J Biol Chem 2005; 280:36334-41; PMID:16118205; http://dx.doi.org/10.1074/jbc.M508660200
- Duarte-Pereira S, Silva SS, Azevedo L, Castro L, Amorim A, Silva RM. NAMPT and NAPRT1: novel polymorphisms and distribution of variants between normal tissues and tumor samples. Sci Rep 2014; 4:6311; PMID:25201160; http://dx.doi.org/10.1038/srep06311
- Rongvaux A, Galli M, Denanglaire S, Van Gool F, Dreze PL, Szpirer C, Bureau F, Andris F, Leo O. Nicotinamide phosphoribosyl transferase/pre-B cell colony-enhancing factor/visfatin is required for lymphocyte development and cellular resistance to genotoxic stress. J Immunol 2008; 181:4685-95; PMID:18802071; http://dx.doi.org/10.4049/jimmunol.181.7.4685
- Yang H, Yang T, Baur JA, Perez E, Matsui T, Carmona JJ, Lamming DW, Souza-Pinto NC, Bohr VA, Rosenzweig A, et al. Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell 2007; 130:1095-107; PMID:17889652; http://dx.doi.org/10.1016/j.cell.2007.07.035
- Mori V, Amici A, Mazzola F, Di Stefano M, Conforti L, Magni G, Ruggieri S, Raffaelli N, Orsomando G. Metabolic Profiling of Alternative NAD Biosynthetic Routes in Mouse Tissues. PLoS One 2014; 9:e113939; PMID:25423279; http://dx.doi.org/10.1371/journal.pone.0113939
- Bieganowski P, Brenner C. Discoveries of nicotinamide riboside as a nutrient and conserved NRK genes establish a Preiss-Handler independent route to NAD+ in fungi and humans. Cell 2004; 117:495-502; PMID:15137942; http://dx.doi.org/10.1016/S0092-8674(04)00416-7
- Hara N, Yamada K, Shibata T, Osago H, Hashimoto T, Tsuchiya M. Elevation of cellular NAD levels by nicotinic acid and involvement of nicotinic acid phosphoribosyltransferase in human cells. J Biol Chem 2007; 282:24574-82; PMID:17604275; http://dx.doi.org/10.1074/jbc.M610357200
- Chang YH, Chang DM, Lin KC, Shin SJ, Lee YJ. Visfatin in overweight/obesity, type 2 diabetes mellitus, insulin resistance, metabolic syndrome and cardiovascular diseases: a meta-analysis and systemic review. Diabetes Metab Res Rev 2011; 27:515-27; PMID:21484978; http://dx.doi.org/10.1002/dmrr.1201
- Revollo JR, Korner A, Mills KF, Satoh A, Wang T, Garten A, Dasgupta B, Sasaki Y, Wolberger C, Townsend RR, et al. Nampt/PBEF/Visfatin regulates insulin secretion in beta cells as a systemic NAD biosynthetic enzyme. Cell Metab 2007; 6:363-75; PMID:17983582; http://dx.doi.org/10.1016/j.cmet.2007.09.003
- Hara N, Yamada K, Shibata T, Osago H, Tsuchiya M. Nicotinamide phosphoribosyltransferase/visfatin does not catalyze nicotinamide mononucleotide formation in blood plasma. PLoS One 2011; 6:e22781; PMID:21826208; http://dx.doi.org/10.1371/journal.pone.0022781
- Luo X, Kraus WL. On PAR with PARP: cellular stress signaling through poly(ADP-ribose) and PARP-1. Genes Dev 2012; 26:417-32; PMID:22391446; http://dx.doi.org/10.1101/gad.183509.111
- D'Amours D, Desnoyers S, D'Silva I, Poirier GG. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem J 1999; 342(Pt 2):249-68; PMID:10455009; http://dx.doi.org/10.1042/0264-6021:3420249
- Hassa PO, Haenni SS, Elser M, Hottiger MO. Nuclear ADP-ribosylation reactions in mammalian cells: where are we today and where are we going? Microbiol Mol Biol Rev 2006; 70:789-829; PMID:16959969; http://dx.doi.org/10.1128/MMBR.00040-05
- Li M, Yu X. The role of poly(ADP-ribosyl)ation in DNA damage response and cancer chemotherapy. Oncogene 2015: 34(26):3349-56; PMID:25220415
- Kozako T, Suzuki T, Yoshimitsu M, Arima N, Honda SI, Soeda S. Anticancer agents targeted to sirtuins. Molecules 2014; 19:20295-313; PMID:25486244; http://dx.doi.org/10.3390/molecules191220295
- Imai S, Guarente L. NAD+ and sirtuins in aging and disease. Trends Cell Biol 2014; 24:464-71; PMID:24786309; http://dx.doi.org/10.1016/j.tcb.2014.04.002
- Malavasi F, Deaglio S, Funaro A, Ferrero E, Horenstein AL, Ortolan E, Vaisitti T, Aydin S. Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology. Physiol Rev 2008; 88:841-86; PMID:18626062; http://dx.doi.org/10.1152/physrev.00035.2007
- Wei W, Graeff R, Yue J. Roles and mechanisms of the CD38/cyclic adenosine diphosphate ribose/Ca(2+) signaling pathway. World J Biol Chem 2014; 5:58-67; PMID:24600514; http://dx.doi.org/10.4331/wjbc.v5.i1.58
- Damle RN, Wasil T, Fais F, Ghiotto F, Valetto A, Allen SL, Buchbinder A, Budman D, Dittmar K, Kolitz J, et al. Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. Blood 1999; 94:1840-7; PMID:10477712
- Leo R, Boeker M, Peest D, Hein R, Bartl R, Gessner JE, Selbach J, Wacker G, Deicher H. Multiparameter analyses of normal and malignant human plasma cells: CD38++, CD56+, CD54+, cIg+ is the common phenotype of myeloma cells. Ann Hematol 1992; 64:132-9; PMID:1373957; http://dx.doi.org/10.1007/BF01697400
- Young GS, Choleris E, Lund FE, Kirkland JB. Decreased cADPR and increased NAD+ in the Cd38−/− mouse. Biochem Biophys Res Commun 2006; 346:188-92; PMID:16750163; http://dx.doi.org/10.1016/j.bbrc.2006.05.100
- Howard M, Grimaldi JC, Bazan JF, Lund FE, Santos-Argumedo L, Parkhouse RM, Walseth TF, Lee HC. Formation and hydrolysis of cyclic ADP-ribose catalyzed by lymphocyte antigen CD38. Science 1993; 262:1056-9; PMID:8235624; http://dx.doi.org/10.1126/science.8235624
- Spychala J. Tumor-promoting functions of adenosine. Pharmacol Ther 2000; 87:161-73; PMID:11007998; http://dx.doi.org/10.1016/S0163-7258(00)00053-X
- Garavaglia S, Bruzzone S, Cassani C, Canella L, Allegrone G, Sturla L, Mannino E, Millo E, De Flora A, Rizzi M. The high-resolution crystal structure of periplasmic Haemophilus influenzae NAD nucleotidase reveals a novel enzymatic function of human CD73 related to NAD metabolism. Biochem J 2012; 441:131-41; PMID:21933152; http://dx.doi.org/10.1042/BJ20111263
- Lang R, Yagar EF, Eggers R, Hofmann T. Quantitative investigation of trigonelline, nicotinic acid, and nicotinamide in foods, urine, and plasma by means of LC-MS/MS and stable isotope dilution analysis. J Agric Food Chem 2008; 56:11114-21; PMID:19007232; http://dx.doi.org/10.1021/jf802838s
- Bruzzone S, Guida L, Zocchi E, Franco L, De Flora A. Connexin 43 hemi channels mediate Ca2+-regulated transmembrane NAD+ fluxes in intact cells. FASEB J 2001; 15:10-2; PMID:11099492
- Stein LR, Imai S. The dynamic regulation of NAD metabolism in mitochondria. Trends Endocrinol Metab 2012; 23:420-8; PMID:22819213; http://dx.doi.org/10.1016/j.tem.2012.06.005
- Dolle C, Rack JG, Ziegler M. NAD and ADP-ribose metabolism in mitochondria. FEBS J 2013; 280:3530-41; PMID:23617329; http://dx.doi.org/10.1111/febs.12304
- Nikiforov A, Dolle C, Niere M, Ziegler M. Pathways and subcellular compartmentation of NAD biosynthesis in human cells: from entry of extracellular precursors to mitochondrial NAD generation. J Biol Chem 2011; 286:21767-78; PMID:21504897; http://dx.doi.org/10.1074/jbc.M110.213298
- Van Beijnum JR, Moerkerk PT, Gerbers AJ, De Bruine AP, Arends JW, Hoogenboom HR, Hufton SE. Target validation for genomics using peptide-specific phage antibodies: a study of five gene products overexpressed in colorectal cancer. Int J Cancer 2002; 101:118-27; PMID:12209988; http://dx.doi.org/10.1002/ijc.10584
- Shackelford RE, Bui MM, Coppola D, Hakam A. Over-expression of nicotinamide phosphoribosyltransferase in ovarian cancers. Int J Clin Exp Pathol 2010; 3:522-7; PMID:20606733
- Wang B, Hasan MK, Alvarado E, Yuan H, Wu H, Chen WY. NAMPT overexpression in prostate cancer and its contribution to tumor cell survival and stress response. Oncogene 2011; 30:907-21; PMID:20956937; http://dx.doi.org/10.1038/onc.2010.468
- Reddy PS, Umesh S, Thota B, Tandon A, Pandey P, Hegde AS, Balasubramaniam A, Chandramouli BA, Santosh V, Rao MR, et al. PBEF1/NAmPRTase/Visfatin: a potential malignant astrocytoma/glioblastoma serum marker with prognostic value. Cancer Biol Ther 2008; 7:663-8; PMID:18728403; http://dx.doi.org/10.4161/cbt.7.5.5663
- Maldi E, Travelli C, Caldarelli A, Agazzone N, Cintura S, Galli U, Scatolini M, Ostano P, Miglino B, Chiorino G, et al. Nicotinamide phosphoribosyltransferase (NAMPT) is over-expressed in melanoma lesions. Pigment Cell Melanoma Res 2013; 26:144-6; PMID:23051650; http://dx.doi.org/10.1111/pcmr.12037
- Olesen UH, Hastrup N, Sehested M. Expression patterns of nicotinamide phosphoribosyltransferase and nicotinic acid phosphoribosyltransferase in human malignant lymphomas. APMIS 2011; 119:296-303; PMID:21492230; http://dx.doi.org/10.1111/j.1600-0463.2011.02733.x
- Bi TQ, Che XM, Liao XH, Zhang DJ, Long HL, Li HJ, Zhao W. Overexpression of Nampt in gastric cancer and chemopotentiating effects of the Nampt inhibitor FK866 in combination with fluorouracil. Oncol Rep 2011; 26:1251-7; PMID:21743967
- Tian W, Zhu Y, Wang Y, Teng F, Zhang H, Liu G, Ma X, Sun D, Rohan T, Xue F. Visfatin, a potential biomarker and prognostic factor for endometrial cancer. Gynecol Oncol 2013; 129:505-12; PMID:23438672; http://dx.doi.org/10.1016/j.ygyno.2013.02.022
- Menssen A, Hydbring P, Kapelle K, Vervoorts J, Diebold J, Luscher B, Larsson LG, Hermeking H. The c-MYC oncoprotein, the NAMPT enzyme, the SIRT1-inhibitor DBC1, and the SIRT1 deacetylase form a positive feedback loop. Proc Natl Acad Sci U S A 2012; 109:E187-96; PMID:22190494; http://dx.doi.org/10.1073/pnas.1105304109
- Cagnetta A, Cea M, Calimeri T, Acharya C, Fulciniti M, Tai YT, Hideshima T, Chauhan D, Zhong MY, Patrone F, et al. Intracellular NAD(+) depletion enhances bortezomib-induced anti-myeloma activity. Blood 2013; 122:1243-55; PMID:23823317; http://dx.doi.org/10.1182/blood-2013-02-483511
- Hasmann M, Schemainda I. FK866, a highly specific noncompetitive inhibitor of nicotinamide phosphoribosyltransferase, represents a novel mechanism for induction of tumor cell apoptosis. Cancer Res 2003; 63:7436-42; PMID:14612543
- Watson M, Roulston A, Belec L, Billot X, Marcellus R, Bedard D, Bernier C, Branchaud S, Chan H, Dairi K, et al. The small molecule GMX1778 is a potent inhibitor of NAD+ biosynthesis: strategy for enhanced therapy in nicotinic acid phosphoribosyltransferase 1-deficient tumors. Mol Cell Biol 2009; 29:5872-88; PMID:19703994; http://dx.doi.org/10.1128/MCB.00112-09
- Chini CC, Guerrico AM, Nin V, Camacho-Pereira J, Escande C, Barbosa MT, Chini EN. Targeting of NAD metabolism in pancreatic cancer cells: potential novel therapy for pancreatic tumors. Clin Cancer Res 2014; 20:120-30; PMID:24025713; http://dx.doi.org/10.1158/1078-0432.CCR-13-0150
- Xiao Y, Elkins K, Durieux JK, Lee L, Oeh J, Yang LX, Liang X, DelNagro C, Tremayne J, Kwong M, et al. Dependence of tumor cell lines and patient-derived tumors on the NAD salvage pathway renders them sensitive to NAMPT inhibition with GNE-618. Neoplasia 2013; 15:1151-60; PMID:24204194; http://dx.doi.org/10.1593/neo.131304
- Beauparlant P, Bedard D, Bernier C, Chan H, Gilbert K, Goulet D, Gratton MO, Lavoie M, Roulston A, Turcotte E, et al. Preclinical development of the nicotinamide phosphoribosyl transferase inhibitor prodrug GMX1777. Anticancer Drugs 2009; 20:346-54; PMID:19369827; http://dx.doi.org/10.1097/CAD.0b013e3283287c20
- Chan M, Gravel M, Bramoulle A, Bridon G, Avizonis D, Shore GC, Roulston A. Synergy between the NAMPT Inhibitor GMX1777(8) and pemetrexed in non-small cell lung cancer cells is mediated by PARP activation and enhanced NAD consumption. Cancer Res 2014; 74:5948-54; PMID:25145669; http://dx.doi.org/10.1158/0008-5472.CAN-14-0809
- Olesen UH, Thougaard AV, Jensen PB, Sehested M. A preclinical study on the rescue of normal tissue by nicotinic acid in high-dose treatment with APO866, a specific nicotinamide phosphoribosyltransferase inhibitor. Mol Cancer Ther 2010; 9:1609-17; PMID:20515945; http://dx.doi.org/10.1158/1535-7163.MCT-09-1130
- O'Brien T, Oeh J, Xiao Y, Liang X, Vanderbilt A, Qin A, Yang L, Lee LB, Ly J, Cosino E, et al. Supplementation of nicotinic acid with NAMPT inhibitors results in loss of in vivo efficacy in NAPRT1-deficient tumor models. Neoplasia 2013; 15:1314-29; PMID:24403854; http://dx.doi.org/10.1593/neo.131718
- Adams DJ, Ito D, Rees MG, Seashore-Ludlow B, Puyang X, Ramos AH, Cheah JH, Clemons PA, Warmuth M, Zhu P, et al. NAMPT is the cellular target of STF-31-like small-molecule probes. ACS Chem Biol 2014; 9:2247-54; PMID:25058389; http://dx.doi.org/10.1021/cb500347p
- Binderup E, Bjorkling F, Hjarnaa PV, Latini S, Baltzer B, Carlsen M, Binderup L. EB1627: a soluble prodrug of the potent anticancer cyanoguanidine CHS828. Bioorg Med Chem Lett 2005; 15:2491-4; PMID:15863303; http://dx.doi.org/10.1016/j.bmcl.2005.03.064
- Zheng X, Bauer P, Baumeister T, Buckmelter AJ, Caligiuri M, Clodfelter KH, Han B, Ho YC, Kley N, Lin J, et al. Structure-based discovery of novel amide-containing nicotinamide phosphoribosyltransferase (nampt) inhibitors. J Med Chem 2013; 56:6413-33; PMID:23859118; http://dx.doi.org/10.1021/jm4008664
- Zheng X, Bair KW, Bauer P, Baumeister T, Bowman KK, Buckmelter AJ, Caligiuri M, Clodfelter KH, Feng Y, Han B, et al. Identification of amides derived from 1H-pyrazolo[3,4-b]pyridine-5-carboxylic acid as potent inhibitors of human nicotinamide phosphoribosyltransferase (NAMPT). Bioorg Med Chem Lett 2013; 23:5488-97; PMID:24021463; http://dx.doi.org/10.1016/j.bmcl.2013.08.074
- Christensen MK, Erichsen KD, Olesen UH, Tjornelund J, Fristrup P, Thougaard A, Nielsen SJ, Sehested M, Jensen PB, Loza E, et al. Nicotinamide phosphoribosyltransferase inhibitors, design, preparation, and structure-activity relationship. J Med Chem 2013; 56:9071-88; PMID:24164086; http://dx.doi.org/10.1021/jm4009949
- Sampath D, Zabka TS, Misner DL, O'Brien T, Dragovich PS. Inhibition of nicotinamide phosphoribosyltransferase (NAMPT) as a therapeutic strategy in cancer. Pharmacol Ther 2015; 51:16-31; PMID:25709099
- Olesen UH, Christensen MK, Björkling F, Jäättelä M, Jensen PB, Sehested M, Nielsen SJ. Anticancer agent CHS-828 inhibits cellular synthesis of NAD. Biochem Biophys Res Commun 2008; 367:799-804; PMID:18201551; http://dx.doi.org/10.1016/j.bbrc.2008.01.019
- Oh A, Ho YC, Zak M, Liu Y, Chen X, Yuen PW, Zheng X, Liu Y, Dragovich PS, Wang W. Structural and biochemical analyses of the catalysis and potency impact of inhibitor phosphoribosylation by human nicotinamide phosphoribosyltransferase. Chembiochem 2014; 15:1121-30; PMID:24797455; http://dx.doi.org/10.1002/cbic.201402023
- Tan B, Young DA, Lu ZH, Wang T, Meier TI, Shepard RL, Roth K, Zhai Y, Huss K, Kuo MS, et al. Pharmacological inhibition of nicotinamide phosphoribosyltransferase (NAMPT), an enzyme essential for NAD+ biosynthesis, in human cancer cells: metabolic basis and potential clinical implications. J Biol Chem 2013; 288:3500-11; PMID:23239881; http://dx.doi.org/10.1074/jbc.M112.394510
- Tolstikov V, Nikolayev A, Dong S, Zhao G, Kuo MS. Metabolomics analysis of metabolic effects of nNicotinamide phosphoribosyltransferase (NAMPT) inhibition on human cancer cells. PLoS One 2014; 9:e114019; PMID:25486521; http://dx.doi.org/10.1371/journal.pone.0114019
- Del Nagro C, Xiao Y, Rangell L, Reichelt M, O'Brien T. Depletion of the central metabolite NAD leads to oncosis mediated cell death. J Biol Chem 2014; 289:35182-92; PMID:25355314
- Muruganandham M, Alfieri AA, Matei C, Chen Y, Sukenick G, Schemainda I, Hasmann M, Saltz LB, Koutcher JA. Metabolic signatures associated with a NAD synthesis inhibitor-induced tumor apoptosis identified by 1H-decoupled-31P magnetic resonance spectroscopy. Clin Cancer Res 2005; 11:3503-13; PMID:15867253; http://dx.doi.org/10.1158/1078-0432.CCR-04-1399
- Pittelli M, Formentini L, Faraco G, Lapucci A, Rapizzi E, Cialdai F, Romano G, Moneti G, Moroni F, Chiarugi A. Inhibition of nicotinamide phosphoribosyltransferase: cellular bioenergetics reveals a mitochondrial insensitive NAD pool. J Biol Chem 2010; 285:34106-14; PMID:20724478; http://dx.doi.org/10.1074/jbc.M110.136739
- Billington RA, Genazzani AA, Travelli C, Condorelli F. NAD depletion by FK866 induces autophagy. Autophagy 2008; 4:385-7; PMID:18227641; http://dx.doi.org/10.4161/auto.5635
- Kato H, Ito E, Shi W, Alajez NM, Yue S, Lee C, Chan N, Bhogal N, Coackley CL, Vines D, et al. Efficacy of combining GMX1777 with radiation therapy for human head and neck carcinoma. Clin Cancer Res 2010; 16:898-911; PMID:20103674; http://dx.doi.org/10.1158/1078-0432.CCR-09-1945
- Moore Z, Chakrabarti G, Luo X, Ali A, Hu Z, Fattah FJ, Vemireddy R, DeBerardinis RJ, Brekken RA, Boothman DA. NAMPT inhibition sensitizes pancreatic adenocarcinoma cells to tumor-selective, PAR-independent metabolic catastrophe and cell death induced by beta-lapachone. Cell Death Dis 2015; 6:e1599; PMID:25590809; http://dx.doi.org/10.1038/cddis.2014.564
- Plummer R. Poly(ADP-ribose)polymerase (PARP) inhibitors: from bench to bedside. Clin Oncol 2014; 26:250-6; PMID:24602564; http://dx.doi.org/10.1016/j.clon.2014.02.007
- Zerp SF, Vens C, Floot B, Verheij M, van Triest B. NAD(+) depletion by APO866 in combination with radiation in a prostate cancer model, results from an in vitro and in vivo study. Radiother Oncol 2014; 110:348-54; PMID:24412016; http://dx.doi.org/10.1016/j.radonc.2013.10.039
- Goellner EM, Grimme B, Brown AR, Lin YC, Wang XH, Sugrue KF, Mitchell L, Trivedi RN, Tang JB, Sobol RW. Overcoming temozolomide resistance in glioblastoma via dual inhibition of NAD+ biosynthesis and base excision repair. Cancer Res 2011; 71:2308-17; PMID:21406402; http://dx.doi.org/10.1158/0008-5472.CAN-10-3213
- Venkateshaiah SU, Khan S, Ling W, Bam R, Li X, van Rhee F, Usmani S, Barlogie B, Epstein J, Yaccoby S. NAMPT/PBEF1 enzymatic activity is indispensable for myeloma cell growth and osteoclast activity. Exp Hematol 2013; 41:547-57.e2; PMID:23435312; http://dx.doi.org/10.1016/j.exphem.2013.02.008
- Skoge RH, Dolle C, Ziegler M. Regulation of SIRT2-dependent alpha-tubulin deacetylation by cellular NAD levels. DNA Repair (Amst) 2014; 23:33-8; PMID:24814981; http://dx.doi.org/10.1016/j.dnarep.2014.04.011
- Bowlby SC, Thomas MJ, D'Agostino RB, Jr., Kridel SJ. Nicotinamide phosphoribosyl transferase (Nampt) is required for de novo lipogenesis in tumor cells. PLoS One 2012; 7:e40195; PMID:22768255; http://dx.doi.org/10.1371/journal.pone.0040195
- Michels J, Obrist F, Castedo M, Vitale I, Kroemer G. PARP and other prospective targets for poisoning cancer cell metabolism. Biochem Pharmacol 2014; 92:164-71; PMID:25199458; http://dx.doi.org/10.1016/j.bcp.2014.08.026
- Wang W, Elkins K, Oh A, Ho YC, Wu J, Li H, Xiao Y, Kwong M, Coons M, Brillantes B, et al. Structural basis for resistance to diverse classes of NAMPT inhibitors. PLoS One 2014; 9:e109366; PMID:25285661; http://dx.doi.org/10.1371/journal.pone.0109366
- Olesen UH, Petersen JG, Garten A, Kiess W, Yoshino J, Imai S, Christensen MK, Fristrup P, Thougaard AV, Bjorkling F, et al. Target enzyme mutations are the molecular basis for resistance towards pharmacological inhibition of nicotinamide phosphoribosyltransferase. BMC Cancer 2010; 10:677; PMID:21144000; http://dx.doi.org/10.1186/1471-2407-10-677
- Holen K, Saltz LB, Hollywood E, Burk K, Hanauske AR. The pharmacokinetics, toxicities, and biologic effects of FK866, a nicotinamide adenine dinucleotide biosynthesis inhibitor. Invest New Drugs 2008; 26:45-51; PMID:17924057; http://dx.doi.org/10.1007/s10637-007-9083-2
- Hovstadius P, Larsson R, Jonsson E, Skov T, Kissmeyer AM, Krasilnikoff K, Bergh J, Karlsson MO, Lonnebo A, Ahlgren J. A Phase I study of CHS 828 in patients with solid tumor malignancy. Clin Cancer Res 2002; 8:2843-50; PMID:12231525
- Pishvaian MJ, Marshall JL, Hwang JH, Malik SM, He AR, Deeken JF, Kelso CB, Dorsch-Vogel K, Berger MS. A phase 1 trial of GMX1777: An inhibitor of nicotinamide phosphoribosyl transferase (NAMPT). J Clin Oncol 2008; 26:14568
- Ravaud A, Cerny T, Terret C, Wanders J, Bui BN, Hess D, Droz JP, Fumoleau P, Twelves C. Phase I study and pharmacokinetic of CHS-828, a guanidino-containing compound, administered orally as a single dose every 3 weeks in solid tumours: an ECSG/EORTC study. Eur J Cancer 2005; 41:702-7; PMID:15763645; http://dx.doi.org/10.1016/j.ejca.2004.12.023
- von Heideman A, Berglund A, Larsson R, Nygren P. Safety and efficacy of NAD depleting cancer drugs: results of a phase I clinical trial of CHS 828 and overview of published data. Cancer Chemother Pharmacol 2010; 65:1165-72; PMID:19789873; http://dx.doi.org/10.1007/s00280-009-1125-3
- Jacobson EL, Kim H, Kim M, Jacobson MK. Niacin: vitamin and antidyslipidemic drug. Subcell Biochem 2012; 56:37-47; PMID:22116693
- Shames DS, Elkins K, Walter K, Holcomb T, Du P, Mohl D, Xiao Y, Pham T, Haverty PM, Liederer B, et al. Loss of NAPRT1 expression by tumor-specific promoter methylation provides a novel predictive biomarker for NAMPT inhibitors. Clin Cancer Res 2013; 19:6912-23; PMID:24097869; http://dx.doi.org/10.1158/1078-0432.CCR-13-1186
- Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A, et al. Proteomics. Tissue-based map of the human proteome. Science 2015; 347:1260419; PMID:25613900; http://dx.doi.org/10.1126/science.1260419
- Pittelli M, Cavone L, Lapucci A, Oteri C, Felici R, Niccolai E, Amedei A, Chiarugi A. Nicotinamide phosphoribosyltransferase (NAMPT) activity is essential for survival of resting lymphocytes. Immunol Cell Biol 2014; 92:191-9; PMID:24275857; http://dx.doi.org/10.1038/icb.2013.85
- Zabka TS, Singh JT, Dhawan P, Liederer BM, Oeh J, Kauss MA, Xiao Y, Zak M, Lin T, McCray B, et al. Retinal toxicity, in vivo and in vitro, associated with inhibition of nicotinamide phosphoribosyltransferase. Toxicol Sci 2015; 144(1):163-72; PMID:25505128
- Gehrke I, Bouchard ED, Beiggi S, Poeppl AG, Johnston JB, Gibson SB, Banerji V. On-target effect of FK866, a nicotinamide phosphoribosyl transferase inhibitor, by apoptosis-mediated death in chronic lymphocytic leukemia cells. Clin Cancer Res 2014; 20:4861-72; PMID:25172933; http://dx.doi.org/10.1158/1078-0432.CCR-14-0624
- Weeks LD, Fu P, Gerson SL. Uracil-DNA glycosylase expression determines human lung cancer cell sensitivity to pemetrexed. Mol Cancer Ther 2013; 12:2248-60; PMID:23873851; http://dx.doi.org/10.1158/1535-7163.MCT-13-0172
- Barnes DE, Lindahl T. Repair and genetic consequences of endogenous DNA base damage in mammalian cells. Ann Rev Genet 2004; 38:445-76; PMID:15568983; http://dx.doi.org/10.1146/annurev.genet.38.072902.092448
- Tang JB, Goellner EM, Wang XH, Trivedi RN, St Croix CM, Jelezcova E, Svilar D, Brown AR, Sobol RW. Bioenergetic metabolites regulate base excision repair-dependent cell death in response to DNA damage. Mol Cancer Res 2010; 8:67-79; PMID:20068071; http://dx.doi.org/10.1158/1541-7786.MCR-09-0411
- Andrabi SA, Dawson TM, Dawson VL. Mitochondrial and nuclear cross talk in cell death: parthanatos. Ann N Y Acad Sci 2008; 1147:233-41; PMID:19076445; http://dx.doi.org/10.1196/annals.1427.014
- Bajrami I, Kigozi A, Van Weverwijk A, Brough R, Frankum J, Lord CJ, Ashworth A. Synthetic lethality of PARP and NAMPT inhibition in triple-negative breast cancer cells. EMBO Mol Med 2012; 4:1087-96; PMID:22933245; http://dx.doi.org/10.1002/emmm.201201250
- Cea M, Soncini D, Fruscione F, Raffaghello L, Garuti A, Emionite L, Moran E, Magnone M, Zoppoli G, Reverberi D, et al. Synergistic interactions between HDAC and sirtuin inhibitors in human leukemia cells. PLoS One 2011; 6:e22739; PMID:21818379; http://dx.doi.org/10.1371/journal.pone.0022739
- Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, Deck LM, Royer RE, Vander Jagt DL, Semenza GL, Dang CV. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci U S A 2010; 107:2037-42; PMID:20133848; http://dx.doi.org/10.1073/pnas.0914433107
- Zoppoli G, Cea M, Soncini D, Fruscione F, Rudner J, Moran E, Caffa I, Bedognetti D, Motta G, Ghio R, et al. Potent synergistic interaction between the Nampt inhibitor APO866 and the apoptosis activator TRAIL in human leukemia cells. Exp Hematol 2010; 38:979-88; PMID:20696207; http://dx.doi.org/10.1016/j.exphem.2010.07.013
- Nahimana A, Aubry D, Breton CS, Majjigapu SR, Sordat B, Vogel P, Duchosal MA. The anti-lymphoma activity of APO866, an inhibitor of nicotinamide adenine dinucleotide biosynthesis, is potentialized when used in combination with anti-CD20 antibody. Leuk Lymphoma 2014; 55:2141-50; PMID:24283753; http://dx.doi.org/10.3109/10428194.2013.869325
- Pogrebniak A, Schemainda I, Azzam K, Pelka-Fleischer R, Nussler V, Hasmann M. Chemopotentiating effects of a novel NAD biosynthesis inhibitor, FK866, in combination with antineoplastic agents. Eur J Med Res 2006; 11:313-21; PMID:17052966