
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Next-generation sequencing data from human cancers are often difficult to interpret within the context of tumor evolution. We developed a mathematical model describing the accumulation of mutations under neutral evolutionary dynamics and showed that 323/904 cancers (∼30%) from multiple types were consistent with the neutral model of tumor evolution.
Deep sequencing of tumor samples reveals the fraction of cells that harbor individual mutations. These mutations are the result of replication errors during cell division and their frequency distribution is a consequence of each cancer's evolutionary history. In our latest workCitation1 we utilized the distribution of mutant allele frequencies of tumors to infer how individual cancers grew.
The shape of the mutant allele frequency distribution is determined by numerous factors, such as the growth characteristics of a tumor, the mutation rate per division, and, potentially, the presence of differentially growing subclones. In particular, the occurrence of multiple subclones can introduce arbitrary complexity in the clonal composition of the tumor, making interpretation of the mutant allele frequency distribution extremely difficult.
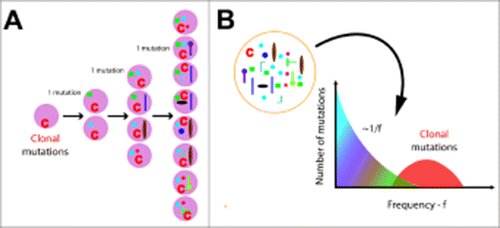
We started from the simplest possible scenario where all cells in the tumor grow at the same rate, a situation where the tumor follows “neutral evolution”, which is the null model for molecular evolution.Citation2,3 Mathematically, this leads to a distribution of mutations that follows a power law. Given a mutation rate μ, a probability β that cells produce 2 surviving offspring, and the frequency of mutations f, our model predicts that the cumulative number of mutations with a frequency greater than f, M(f), is linearly proportional to 1/f,[1]
[1]
Equation Equation[1][1]
[1] has been described previously in the population genetics literature,Citation2,3 but has not to our knowledge been applied to cancer genomes. We tested the prediction of our model on next-generation sequencing data from 904 tumors of many different types. Remarkably, we found that more than one-third of all cases were consistent with this simple model.
Under this neutral model of tumor evolution, the frequency of a mutation in the tumor is a proxy for the time it occurred, as early events are present at a high frequency while later events remain at low frequencies, see also . Mutational timelines can therefore be resolved, as can measurements of the mutation rate in individual cancers. Our mutation rate measurements yielded rates of the order 10−6-10−7 bp/division, which were higher than previous estimates (10−9 bp/divisionCitation4). This is because what we measure is an “effective mutation rate”, μ/β. If there is high cell death in the tumor (low β) the true mutation rate may be much lower. Moreover, we note that this was one of the first attempts to measure mutation rates in vivo in human malignancies, whereas previous measurements were performed in vitro or were speculative.
Deviations from the predictions of equation Equation[1][1]
[1] indicate more complex growth patterns such as the presence of functionally distinct subclones. Ongoing clonal selection in cancer has been the traditional view of tumor evolution;Citation5 however, recent evidence might change this presumption. Multiregion profiling studies in colorectal cancer have found neutral evolution to be consistent with genomic data: using the diversity of methylation patterns as a measure of the age of a tumor, Siegmund et al.Citation6 observed “flat” clonal expansions where the age of samples from opposite parts of the tumor appeared the same, consistent with neutral evolution. More recently, we proposed and validated a “Big Bang” model of colon cancer, where malignancies were characterized by numerous intermixed subclones and lack of stringent selection.Citation7 Our results extended these first observations of neutral evolution, demonstrating that neutral evolution can also explain the intratumor heterogeneity seen in other cancer types. Interestingly, some cancer types seem more prone to neutral evolution than others, indicating that the cellular architecture and local microenvironmental conditions are likely important factors in determining the rules of tumor evolution.
Our measurement of neutrality has the benefit that it only requires a single sample, and can therefore be applied to a large amount of existing data. However, our approach only reveals the evolutionary dynamics within this sample. A “global” inference of neutrality across an entire tumor requires multiple samples from different locations of the same tumor. In a recent study, Ling et al.Citation8 considered a large number of samples from a single hepatocellular carcinoma. They also found that the dynamics conform to a neutrally expanding tumor based on an estimate of the total number of mutations in the tumor. Future work should address the basis of “local” versus “global” neutrality.
Overall, our model predicts that neutral evolution leaves a characteristic signature in the allelic frequency distribution. Identifying this signature in cancer genomes enables us to vastly simplify the interpretation of what appear to be inherently complex and noisy data. Consequently, our work demonstrates how integrating physically informed mathematical models and cancer genomics data can provide new, and perhaps unexpected, insight into these complex data.
Recently, physicists were able to demonstrate the existence of gravitational waves by detecting an incredibly weak signal amidst a noisy dataset.Citation9 This was feasible because scientists knew a priori what the signal of a gravitational wave would look like thanks to Einstein's field equations formulated a century earlier. Cancer genomics at present suffers from the reverse problem: large quantities of data can already be generated but the underlying theory remains underdeveloped. What is lacking is the connection between theoretical and experimental frameworks that would allow us to fully interpret and evaluate the wealth of genomic data. Our latest work is, we hope, a first step in this direction.
Figure 1. Mutations and their frequency determine the history of individual tumors. (A) Mutations label distinct lineages and as a tumor grows the size of these lineages becomes progressively smaller. Here, with one mutation per division and all cells growing at the same rate, the 2 mutations occurring during the first division are each present in 50% of the population, whereas mutations occurring during the final division are present in 12.5%. (B) When sequencing tumor biopsies we measure the frequency f of mutations in the population. Neutral tumor evolution imprints a characteristic 1/f signature in the distribution of subclonal mutant allele frequencies.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- Williams MJ, Werner B, Barnes CP, Graham TA, Sottoriva A. Identification of neutral tumor evolution across cancer types. Nat Genet 2016; 48:3; 238–244; http://dx.doi.org/10.1038/ng.3489
- Durrett R. Population genetics of neutral mutations in exponentially growing cancer cell populations. Ann Appl Probability 2013; 23:230-50; PMID:23471293; http://dx.doi.org/10.1214/11-AAP824
- Griffiths RC, Tavaré S. The age of a mutation in a general coalescent tree. Communications in Statistics. Stochastic Models 1998; 14:273-95; http://dx.doi.org/10.1080/15326349808807471
- Jones S. et al. Comparative lesion sequencing provides insights into tumor evolution. Proc Natl Acad Sci USA 2008; 105:4283-8; PMID:18337506; http://dx.doi.org/10.1073/pnas.0712345105
- Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell 1990; 61:759-67; PMID:2188735; http://dx.doi.org/10.1016/0092-8674(90)90186-I
- Siegmund KD, Marjoram P, Woo Y-J, Tavaré S, Shibata D. Inferring clonal expansion and cancer stem cell dynamics from DNA methylation patterns in colorectal cancers. Proc Natl Acad Sci USA 2009; 106:4828-33; PMID:19261858; http://dx.doi.org/10.1073/pnas.0810276106
- Sottoriva A. et al. A Big Bang model of human colorectal tumor growth. Nat Genet 2015; 47:209-16; PMID:25665006; http://dx.doi.org/10.1038/ng.3214
- Ling S. et al. Extremely high genetic diversity in a single tumor points to prevalence of non-Darwinian cell evolution. Proc Natl Acad Sci USA 2015; 112:E6496-505; PMID:26561581; http://dx.doi.org/10.1073/pnas.1519556112
- Abbott BP. et al. Observation of Gravitational Waves from a Binary Black Hole Merger. Phys Rev Lett 2016; 116:061102; PMID:26918975; http://dx.doi.org/10.1103/PhysRevLett.116.061102