
ABSTRACT
The cornerstone for precision medicine is the development of robust biomarkers that reflect molecular phenotypes and therapeutic vulnerabilities in disease. We recently described Janus kinase-2 (JAK2)-specific inhibition as a therapeutic opportunity in triple negative breast cancers with 9p24 amplification. Here, we comment on this work and discuss the challenges of targeting this amplicon.
KEYWORDS:
Clinical significance of selective 9p24 amplification in cancer
In our recent study, we hypothesized that residual triple negative breast cancers (TNBC) remaining after neoadjuvant chemotherapy harbor targetable alterations responsible for subsequent recurrence.Citation1 We found that 9p24 amplification was associated with poor progression-free survival (PFS) and overall survival (OS). Similar findings were reported by Barrett et al. in a cohort of 41 patients with TNBC, in which presence of the 9p24 amplicon (log2 ratio ≥1) was associated with poor survival.Citation2 One of the genes on this amplicon, Janus kinase-2 (JAK2) appeared to support proliferation and maintenance of cancer stem cell (CSC)-like cells, a population known to be spared by chemotherapy.
The 9p24 region involves at least 4 known cancer-related genes: JAK2, CD274 (also known as PD-L1), PCD1LG2 (also known as PD-L2), and KDM4C. Using functional genomics, Rui et al. evaluated how more than one gene can cooperate within the 9p24 amplicon and demonstrated that JAK2 can modulate the epigenome through KDM4C, a histone demethylase.Citation3 JAK2 can also regulate CD274/PD-L1 expression via the ISRE/IRF1 control element.Citation4 Collectively, these data suggest that 9p24 amplification is an opportunistic genomic event that provides multiple cooperative “drivers.” In our study, we focused on the role of JAK2 in this amplicon.
The 9p24 amplicon exists in a variety of human cancers. Around 30% of classical Hodgkin lymphomas (cHL) harbor this alteration, which is associated with advanced stage, shorter PFS, and remarkable PD-1 blockade activity.Citation5 Approximately 50% of primary mediastinal B-cell lymphomas (PMBCLs) show amplification of 9p24 and share a gene expression profile different from that of other diffuse large B-cell lymphomas. Consistent with our results, cHL and PMBCL tumors with 9p24 amplification benefit from JAK2 inhibition in animal models.Citation6 The efficacy of both JAK2 inhibition and PD-1 immunotherapy in these models support a driver status for multiple amplicon-associated genes. However, our work involved human models engrafted in immunocompromised mice, and thus the impact of this amplicon on PD-1-targeting agents was not tested.
JAK inhibitors in clinical trials
Ruxolitinib (an equipotent JAK1 and JAK2 inhibitor) has become the standard of care for intermediate/high risk myelofibrosis (IMF) and hydroxurea-resistant polycythemia vera (PV). The fact that ruxolitinib exerts a modest effect in cells with JAK2V617F (the oncogenic JAK2-activating mutation present in 95% of PVs and 60% of IMFs) suggests that resistance to therapy often develops in the absence of secondary (acquired) mutations. Moreover, primary endpoints in clinical trials are focused on anti-inflammatory effects (improvement of systemic symptoms and splenomegaly) potentially related to IL-6/JAK1 inhibition. This raises an important question: Does the effect of ruxolitinib in these tumors predominately arise from targeting JAK1 or JAK2?
From a mechanistic point of view, JAK inhibitors can be divided into type I (i.e., those binding to the active kinase in an ATP competitive manner) and type II (those binding to the inactive form of the kinase). Andraos et al. showed that type II inhibition provides a more comprehensive JAK2 blockade, avoiding the activation loop phosphorylation of multiple JAK family members produced by type I inhibitors.Citation7 Thus, in addition to targeting JAK1, type I JAK inhibitors may cause reflexive overactivation during drug concentration troughs. Recently, Meyer et al. reported that the type II JAK2 inhibitor CHZ868 produced rapid normalization of clinical as well as molecular parameters in different JAK2-driven myeloproliferative neoplasia and B-cell leukemia mouse models. The strong apoptotic effect on JAK2V617F and JAK2W515L mutant cells and reduction of the mutant allele burden sharply contrasted with a minimal effect on normal hematopoiesis.Citation8 Together with our data, these findings support selective JAK2 inhibition, which may have enhanced tumor-specific effects while sparing toxicities associated with JAK1 inhibition.
There is strong evidence that the immune microenvironment can shape the outcome of cancer and growing interest in the influence of targeted therapies on the immune response. Type I and type II cytokine receptors are major players in cellular and antibody-mediated immunity and mediate their effects via the JAK-STAT pathway. Thus, evaluation of the impact of specific JAK (1/2/3) inhibition on immune competency will be important. Ruxolitinib has been described as an immunosuppressive compound able to inhibit the IL2-dependent JAK1/JAK3-STAT5a activation required for T helper cell maturation and to impair dendritic cell function. This provides a plausible explanation for the high rate of opportunistic infections in patients enrolled on ruxolitinib trials.Citation9 Recently Gotthardt et al. also revealed that genetic and pharmacologic STAT5 inhibition in natural killer cells can induce angiogenesis and promote tumor progression.Citation10 Thus, JAK2-specific inhibition may also circumvent suppression of antitumor immunity caused by JAK inhibitors with broader activity ().
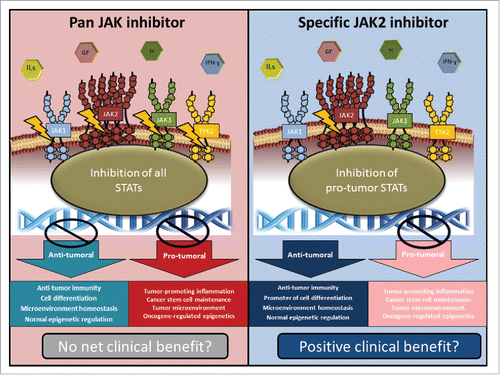
Figure 1. Possible role for selective JAK2 inhibition in JAK2-amplified TNBC. Growth factors (GF), hormones (H), and interleukin cytokines (IL) activate a diversity of JAK family-bound receptors. JAK occupancy on these receptors can dictate the nature of downstream STAT responders. Specific STATs may have pro-tumor or pro-host effects within the tumor. Our data suggest that in JAK2-amplified TNBC, specific JAK2 inhibition may selectively target JAK2-mediated pro-tumor signals without compromising pro-host JAK1/3/TYK signals.
Study limitations
Our work suggests that JAK2 amplification may preferentially affect CSCs, raising the difficulty of elucidating how JAK2 amplification shapes JAK1 and JAK2 signaling in only a subpopulation of tumor cells. Also, the specific downstream (STAT) effectors of JAK2 in TNBCs are not yet clear, although we observed a reduction of STAT5/STAT6 phosphorylation following JAK2-specific inhibition. Surprisingly, we found that STAT3, a common STAT downstream of JAK2, was not affected by JAK2 inhibition but instead relied almost exclusively on JAK1 for activity. Finally, although NVP-BSK-805 effectively targets JAK2amp breast cancer cells, the pharmacologic effect of JAK2 inhibition on stromal cells has yet to be determined.
Despite the striking correlation observed between JAK2amp, poor pathologic response, and shorter overall survival, 9p24 amplification is an infrequent genomic alteration (∼10%) in chemotherapy-selected TNBCs (n = 111). Institutional collaboration will be required to screen a sufficient number of TNBC patients to validate the prognostic and predictive role of 9p24 amplification in breast cancer. Furthermore, these efforts will require substantial efforts in precision medicine to determine whether JAK2amp TNBCs specifically and selectively require JAK2 inhibition to overcome chemotherapeutic resistance.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- Balko JM, Schwarz LJ, Luo N, Estrada MV, Giltnane JM, Davila-Gonzalez D, et al. Triple-negative breast cancers with amplification of JAK2 at the 9p24 locus demonstrate JAK2-specific dependence. Sci Transl Med 2016 Apr 13; 8(334):334ra53; PMID:27075627; http://dx.doi.org/10.1126/scitranslmed.aad3001
- Barrett MT, Anderson KS, Lenkiewicz E, Andreozzi M, Cunliffe HE, Klassen CL, et al. Genomic amplification of 9p24.1 targeting JAK2, PD-L1, and PD-L2 is enriched in high-risk triple negative breast cancer. Oncotarget 2015 Sep 22; 6(28):26483-93; PMID:26317899; http://dx.doi.org/10.18632/oncotarget.4494
- Rui L, Emre NC, Kruhlak MJ, Chung HJ, Steidl C, Slack G, et al. Cooperative epigenetic modulation by cancer amplicon genes. Cancer Cell. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov't]. 2010 Dec 14; 18(6):590-605; PMID:21156283
- Green MR, Monti S, Rodig SJ, Juszczynski P, Currie T, O'Donnell E, et al. Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood 2010 Oct 28; 116(17):3268-77;PMID:20628145; http://dx.doi.org/10.1182/blood-2010-05-282780
- Roemer MG, Advani RH, Ligon AH, Natkunam Y, Redd RA, Homer H, et al. PD-L1 and PD-L2 Genetic Alterations Define Classical Hodgkin Lymphoma and Predict Outcome. J Clin Oncol 2016 Apr 11; PMID:2706908; http://dx.doi.org/10.1200/JCO.2016.66.4482
- Hao Y, Chapuy B, Monti S, Sun HH, Rodig SJ, Shipp MA. Selective JAK2 inhibition specifically decreases Hodgkin lymphoma and mediastinal large B-cell lymphoma growth in vitro and in vivo. Clin Cancer Res [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov't]. 2014 May 15; 20(10):2674-83; PMID:24610827; http://dx.doi.org/10.1158/1078-0432.CCR-13-3007
- Andraos R, Qian Z, Bonenfant D, Rubert J, Vangrevelinghe E, Scheufler C, et al. Modulation of activation-loop phosphorylation by JAK inhibitors is binding mode dependent. Cancer Discov 2012 Jun; 2(6):512-23; PMID:22684457; http://dx.doi.org/10.1158/2159-8290.CD-11-0324
- Meyer SC, Keller MD, Chiu S, Koppikar P, Guryanova OA, Rapaport F, et al. CHZ868, a Type II JAK2 Inhibitor, Reverses Type I JAK Inhibitor Persistence and Demonstrates Efficacy in Myeloproliferative Neoplasms. Cancer Cell [Research Support, N.I.H., Extramural]. 2015 Jul 13; 28(1):15-28; PMID:26175413; http://dx.doi.org/10.1016/j.ccell.2015.06.006
- Heine A, Brossart P, Wolf D. Ruxolitinib is a potent immunosuppressive compound: is it time for anti-infective prophylaxis? Blood [Letter]. 2013 Nov 28; 122(23):3843-4; PMID:24288410; http://dx.doi.org/10.1182/blood-2013-10-531103
- Gotthardt D, Putz EM, Grundschober E, Prchal-Murphy M, Straka E, Kudweis P, et al. STAT5 Is a Key Regulator in NK Cells and Acts as a Molecular Switch from Tumor Surveillance to Tumor Promotion. Cancer Discov 2016 Apr; 6(4):414-29; PMID:26873347; http://dx.doi.org/10.1158/2159-8290.CD-15-0732