
ABSTRACT
The mutations induced by activation-induced cytidine deaminase (AID) trigger antibody diversification but can cause genome instability. We find that AID phosphorylation is an important determinant of “off-target” mutagenesis and identify a drug that increases this activity. These studies demonstrate how dysregulating AID phosphorylation can promote oncogenesis.
During development, lymphocytes induce programmed DNA damage to diversify their immune receptors, a process that threatens genomic integrity. Upon immune activation, B cells enter the germinal center and express activation-induced cytidine deaminase (AID) to initiate Immunoglobulin (IG) somatic hypermutation (SHM) and class switch recombination (CSR). During SHM, AID introduces point mutations at the IG variable region to evolve antibody affinity. During CSR, AID creates double strand breaks (DSBs) at the Immunoglobulin heavy (IGH) switch regions triggering recombination. Although predominantly localized to the IG loci, AID is also promiscuously associated with transcriptionally active regions throughout the genome. This includes recurrent chromosome translocation breakpoint partners, such as BCL6 and MYC.Citation1 As the vast majority of lymphomas originate from post-germinal center B cells, AID mutagenesis carries great risk. We have previously shown that deregulated AID activity could result in oncogenic lesions.Citation2 Thus, defining the factors that influence off-target AID activity is critical to understanding lymphomagenesis.
Previous studies demonstrated that phosphorylation regulates AID activity at Ig genes. Biochemical analysis revealed that AID was phosphorylated on serine 38 (pS38)Citation3 and that pS38 levels are high at Igh genes.Citation4 We assessed the role of this modification in B cells isolated from mice with an AID-S38 to alanine (S38A) substitution. AID-S38A was expressed and enzymatically active, but CSR and SHM were deficient.Citation5 Our most recent study assessed the role of AID-pS38 in generating oncogenic Myc translocations. We found that the AID-S38A substitution did not decrease generation of Myc/Igh translocations during normal CSR.Citation6 Since AID activity at Myc is limiting in translocation generation,Citation7 this assay also served as a proxy for measuring off-target activity and indicated that preventing phosphorylation did not impact AID off-target base-line activity. Further, global DSB formation, measured by analyzing the frequency of γH2AX focus formation, was not appreciably decreased in AID-S38A B cells relative to WT cells.Citation6 These results implied that under non-pathologic conditions AID is minimally phosphorylated at off-target sites.
AID-pS38 levels drive high AID activity at the Igh switch regions. AID is a substrate for the cAMP-dependent protein kinase (PKA).Citation8 During B cell activation, PKA is recruited to the switch regions providing a means to phosphorylate AID and induce local DSBsCitation9 (); however, at non-switch regions the relationship between PKA and AID-pS38 was unclear. Therefore, we treated B cells with several PKA inhibitors but found no significant alterations in total cellular AID-pS38 levels, implying that other kinases phosphorylate AID at non-switch regions. Since the S38 site conforms to the consensus sequence for several kinases, we evaluated several pharmacological inhibitors. We found that the clinically relevant drug, lithium, significantly increased both the total cellular and chromatin-associated levels of AID-pS38. We then assessed the consequence of lithium-induced supra-physiologic pS38 levels on AID activity at the off-target Myc gene.Citation6 We found a significant increase in point mutations and Myc/Igh translocation frequency, as well as a significant increase in genome-wide DSBs. Critically, the effects of lithium were not observed in AID-S38A B cells, demonstrating they were phosphorylation specific. Overall, these studies demonstrated that AID phospho-regulation exerts a level of control beyond AID association.
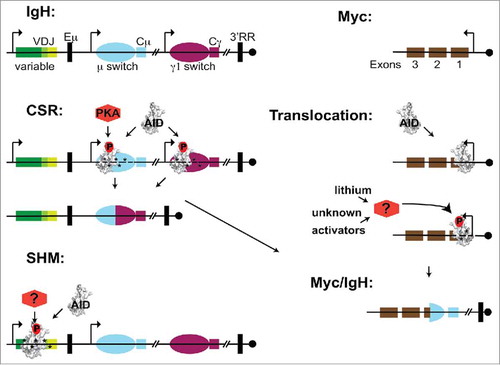
Figure 1. The danger of dysregulated activation-induced cytidine deaminase (AID) phosphorylation. Schematic of the Immunoglobulin heavy (Igh) locus: a recombined V(D)J and constant region exons (colored boxes), non-coding switch regions (ovals), regulatory elements (black boxes) including mu enhancer, downstream enhancers and 3′ regulatory region. The Myc exons (brown boxes) include non-coding exon 1 and centromere orientation (circle) is shown. (B) During class switch recombination (CSR), AID prominently associates with Igh switch regions. However, AID association is promiscuous and occurs at transcriptionally active genes across the genome such as Myc. Phosphorylation on AID serine 38 (AID-pS38) depicted as (red-P), induces mutator activity (*) which cause double strand breaks (DSBs) (gaps) that serve as recombination substrates between isotype switch regions. AID-S38 is a target site for the ubiquitously expressed cAMP-dependent protein kinase (PKA), which is recruited to the Igh switch regions providing a mechanism to induce pS38 and AID activity there. Normally, AID associated with off-target genes normally has minimal AID-pS38 levels. Lithium activation of an unknown kinase pathway induces AID-pS38 levels at off-target sites such as Myc. This results in mutation and DSBs that serve as translocation substrates between Myc and Igh regions. The example depicts a translocation into Exon 1 that brings the coding exons of Myc under the influence of Igh regulatory elements (black boxes) driving overexpression. For somatic hypermutation (SHM), neither the AID targeting mechanism nor the kinase that phosphorylates AID at the Igh variable regions have been defined.
Lithium is a widely prescribed mood stabilizing medication. Its effect on AID-pS38 was surprising, and suggested that patients treated with lithium might be at greater risk for lymphoma. However, no evidence supports this, perhaps because lithium also has anti-tumorigenic properties. Lithium is pleiotropic and affected signaling pathways are numerous. In the future, it will be important to define the pathways and kinases that directly impact AID-pS38. This would facilitate identifying pathologic conditions that promote B cell tumorigenesis through inappropriate AID activity. In turn, this could guide lymphoma prevention/intervention strategies. AID is chronically expressed in a number of B cell cancers including chronic lymphocytic leukemia (CLL) and chronic myeloid leukemia, where it facilitates tumor resistance and clonal evolution. Alternatively, AID expression might be harnessed for anti-tumor effects. The homologous recombination (HR) pathway is critical for preventing the toxic accumulation of AID-induced DSBs. Pharmacological attenuation of HR leads to AID-dependent cytotoxicity in B cells and this has been proposed as a therapeutic strategy for CLL.Citation10 We posit that activating supra-physiologic pS38 levels could enhance the efficacy of such therapeutic intervention.
Author information
The authors declare no competing financial interests.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Additional information
Funding
References
- Casellas R, Basu U, Yewdell WT, Chaudhuri J, Robbiani DF, Di Noia JM. Mutations, kataegis and translocations in B cells: understanding AID promiscuous activity. Nat Rev Immunol. 2016;16(3):164–76. doi:10.1038/nri.2016.2. PMID:26898111.
- Gazumyan A, Timachova K, Yuen G, Siden E, Di Virgilio M, Woo EM, Chait BT, Reina San-Martin B, Nussenzweig MC, McBride KM. Amino-terminal phosphorylation of activation-induced cytidine deaminase suppresses c-myc/IgH translocation. Mol Cell Biol. 2011;31(3):442–9. doi:10.1128/MCB.00349-10. PMID:21135131.
- McBride KM, Gazumyan A, Woo EM, Barreto VM, Robbiani DF, Chait BT, Nussenzweig MC. Regulation of hypermutation by activation-induced cytidine deaminase phosphorylation. Proc Natl Acad Sci U S A. 2006;103(23):8798–803. doi:10.1073/pnas.0603272103. PMID:16723391.
- Vuong BQ, Lee M, Kabir S, Irimia C, Macchiarulo S, McKnight GS, Chaudhuri J. Specific recruitment of protein kinase A to the immunoglobulin locus regulates class-switch recombination. Nat Immunol. 2009;10(4):420–6. doi:10.1038/ni.1708. PMID:19234474.
- McBride KM, Gazumyan A, Woo EM, Schwickert TA, Chait BT, Nussenzweig MC. Regulation of class switch recombination and somatic mutation by AID phosphorylation. J Exp Med. 2008;205(11):2585–94. doi:10.1084/jem.20081319. PMID:18838546.
- Mu Y, Zelazowska MA, McBride KM. Phosphorylation promotes activation-induced cytidine deaminase activity at the Myc oncogene. J Exp Med. 2017;214(12):3543–3552. doi:10.1084/jem.20170468. PMID:29122947.
- Robbiani DF, Bothmer A, Callen E, Reina-San-Martin B, Dorsett Y, Difilippantonio S, Bolland DJ, Chen HT, Corcoran AE, Nussenzweig A, et al. AID is required for the chromosomal breaks in c-myc that lead to c-myc/IgH translocations. Cell. 2008;135(6):1028–38. doi:10.1016/j.cell.2008.09.062. PMID:19070574.
- Basu U, Chaudhuri J, Alpert C, Dutt S, Ranganath S, Li G, Schrum JP, Manis JP, Alt FW. The AID antibody diversification enzyme is regulated by protein kinase A phosphorylation. Nature. 2005;438(7067):508–11. doi:10.1038/nature04255. PMID:16251902.
- Vuong BQ, Herrick-Reynolds K, Vaidyanathan B, Pucella JN, Ucher AJ, Donghia NM, Gu X, Nicolas L, Nowak U, Rahman N, et al. A DNA break- and phosphorylation-dependent positive feedback loop promotes immunoglobulin class-switch recombination. Nat Immunol. 2013;14(11):1183–9. doi:10.1038/ni.2732. PMID:24097111.
- Hasham MG, Snow KJ, Donghia NM, Branca JA, Lessard MD, Stavnezer J, Shopland LS, Mills KD. Activation-induced cytidine deaminase-initiated off-target DNA breaks are detected and resolved during S phase. J Immunol. 2012;189(5):2374–82. doi:10.4049/jimmunol.1200414. PMID:22826323.