
ABSTRACT
In cancer the activity of cyclin-dependent kinase 4-(CDK4) and cyclin-dependent kinase 6 (CDK6)-cyclin complexes are frequently altered with enhanced CDK6 expression found in hematopoietic malignancies. Our latest findings show a so far unknown role of Cdk6 during oncogene-induced stress and transformation. Therein Cdk6 antagonizes p53 responses and subsequently shapes the critical decision between survival and apoptosis in pre-leukemic cells.
Keywords:
Cyclin-dependent kinase 6 (CDK6) in concert with its close homolog cyclin-dependent kinase 4 (CDK4) enables the progression of cells through the early G1 phase of cell cycle by binding to D type cyclins. The CDK4/6-Cyclin D complexes phosphorylate members of the retinoblastoma (RB) protein family thereby releasing E2F transcription factors from RB-mediated inhibition.Citation1 In cancer the activity of CDK4- and CDK6-Cyclin complexes are frequently altered with enhanced CDK6 expression being common in hematopoietic malignancies. Studies in acute lymphoid leukemia (ALL) and acute myeloid leukemia (AML) identified a novel role for CDK6 as a transcriptional regulator, which is not shared by CDK4. As such CDK6 regulates key genes involved in survival, proliferation and angiogenesis including vascular endothelial growth factor A (VEGFA) or fms related tyrosine kinase 3 (FLT3).Citation2–Citation6
Our newest findings put Cdk6 in a yet different context and show that the key function of Cdk6 during oncogene-induced stress and transformation is to antagonize p53 responses. In response to BCR-ABL, lymphoid cells lacking Cdk6 do establish growth-factor independent colonies but are severely impaired when it comes to the formation of stable cell lines. This inability to immortalize was overcome by mutating or deleting tumor protein p53 (Trp53, best known as p53). Any Cdk6-deficient leukemic cell line obtained displayed a mutation in the DNA binding domain of p53. Similarly, lymphocytes expressing a kinase-dead mutant of Cdk6 (Cdk6K43M) were forced to mutate p53 to form leukemic cell lines. This indicates the necessity of Cdk6 kinase activity to antagonize p53 functions during oncogene-induced immortalization.Citation7 The Cdk6 induced transcriptional response is complex and includes a plethora of genes that balance p53-induced pro-apoptotic responses in different ways. We identified the genes encoding for protein arginine methyltransferase 5 (Prmt5) and transformed mouse 3T3 cell double minute 4 (Mdm4) under the transcriptional control of Cdk6. PRMT5 has been shown to directly interact with and methylate p53, thereby suppressing its pro-apoptotic activity.Citation8 Likewise, the p53-interactor MDM4 is considered a major antagonist of its tumor-suppressive functions.
Our findings in the murine system were reflected in human hematopoietic neoplasias where we verified the existence of an overlapping set of CDK6-regulated genes antagonizing p53. Importantly we observed a higher incidence of p53 mutations inversely correlated to CDK6 expression levels in patient cohorts suffering from haematological malignancies including AML, ALL and MDS (myelodysplastic syndrome).Citation7
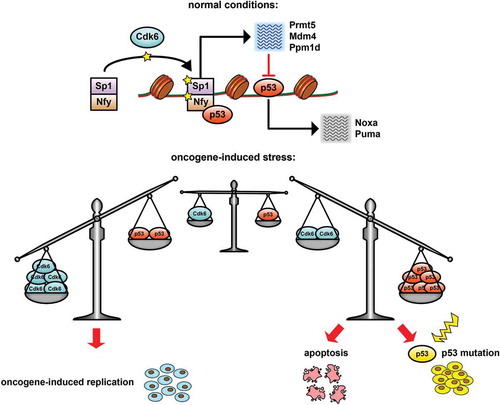
Chromatin immunoprecipitation sequencing (ChIP-seq) for Cdk6 and p53 uncovered a significant peak overlap in the promoter regions of p53 antagonizing genes. At these promoters Cdk6 and p53 DNA binding is predominantly mediated via the adapter proteins trans-acting transcription factor 1 (Sp1) and nuclear transcription factor Y (Nfy). We demonstrated that Cdk6 phosphorylates Nfy and Sp1 at chromatin and propose that this phosphorylation paves the way for p53-binding and subsequent induction of a negative feedback loop limiting its own pro-apoptotic activity. In the absence of Cdk6, chromatin-bound Sp1 and Nfy cannot be efficiently phosphorylated and the pro-apoptotic effects of p53 dominate.Citation7 In response to oncogenic stress, Cdk6 thus tips the scale towards the survival of pre-leukemic clones and supports the emergence and outgrowth of immortalized cell lines (). It effectively plays the part of the emperor in ancient Rome, who indicated by means of his thumb whether a defeated gladiator should be killed or allowed to live.
Figure 1. Key function of cyclin-dependent kinase 6 (Cdk6) during oncogene-induced stress and transformation. Cdk6 phosphorylates nuclear transcription factor Y (Nfy) and trans-acting transcription factor 1 (Sp1) allowing for the p53 (Trp53, best known as p53)- driven transcriptional induction of p53-antagonists including protein arginine methyltransferase 5 (Prmt5), transformed mouse 3T3 cell double minute 4 (Mdm4) and protein phosphatase 1D magnesium-dependent, delta isoform (Ppm1d). This negative feedback loop interferes with p53’s ability to induce pro-apoptotic factors such as phorbol-12-myristate-13-acetate-induced protein 1 (Pmaip1, best known as Noxa) and Bcl2 binding component 3 (Bbc3, best known as Puma). To survive oncogene-induced stress, cells depend on a delicate balance between Cdk6 and p53. Enhanced Cdk6 expression favours oncogene-induced replication and survival, whereas reduced Cdk6 levels tip the scale towards cell death. In the absence of Cdk6, mutating or deleting tumor protein p53 represents a possibility to overcome this road block to immortalization.
The fact that kinase activity of Cdk6 is required to antagonize p53 responses may be seen as potential curse or as blessing. Currently available CDK4/6 kinase inhibitors, such as Palbociclib, have been approved by the US Food and Drug Administration (FDA) for therapeutic use in hormone receptor-positive breast cancer and are in clinical trials for a wide range of tumors including AML.Citation3 Our findings indicate that Palbociclib may enhance p53 induced pro-apoptotic responses upon combination with cytostatic drugs. However, at the flip side of the coin they illustrate that CDK6 kinase inhibition may provoke the outgrowth of p53 mutated sub clones. Potential threats could be prevented by smart drug combinations that immediately eliminate emerging p53 mutated subclones.
Our study also provides a rational for the development of novel drugs. Firstly, we propose that a specific CDK6 kinase inhibitor, which does not impair the activity of CDK4, might be meaningful and advantageous. Many cytostatic drugs work better in proliferating than in quiescent cells – if it is possible to target CDK4 and CDK6 individually, one might block the transcriptional responses by CDK6 with minor impact on cell cycle progression. This may render the cells more vulnerable to p53 activating chemotherapy while ablating p53 antagonistic responses. One option is degrading CDK6 – that would eliminate transcriptional responses that are regulated in a CDK6 kinase – dependent as well as independent manner. This is in particular meaningful for the activation of leukemic stem cell functions that depend on CDK6 but do not require CDK6 kinase activity. One strategy is ligand-induced target protein destabilization which enables degradation of a protein through ligation of a selective small molecule to thalidomide which allows binding to the E3 ligase cereblon (CRBN), a component of a cullin-RING ubiquitin ligase (CRL) complex. Ubiquitination of the protein of interest is followed by proteasomal degradation. The fact that specific degraders for Cyclin-dependent kinase 9 (CDK9) have recently been developed is promising that a similar endeavour may be successful for CDK6.Citation9,Citation10
Alternatively it is attractive to speculate that the ablation of stress-induced CDK6 up-regulation represents an alternative route of targeting p53 feed-back loops to favour p53 induced apoptosis. So far it is not understood how the concomitant upregulation of CDK6 and p53 during oncogene – induced stress is regulated and which signals and signalling networks induce CDK6.
Thereby our study is not the end but the beginning of our journey to develop novel targeting opportunities for CDK6.
Additional information
Funding
References
- Classon M, Harlow E. The retinoblastoma tumour suppressor in development and cancer. Nat Rev Cancer. 2002;2(12):910–917. doi:10.1038/nrc950.
- Scheicher R, Hoelbl-Kovacic A, Bellutti F, Tigan A-S, Prchal-Murphy M, Heller G, Schneckenleithner C, Salazar-Roa M, Ochbauer-Uller S, Zuber J, et al. CDK6 as a key regulator of hematopoietic and leukemic stem cell activation. Blood. 2015; 125(1): 90–101. doi:10.1182/blood-2014-06.
- Uras IZ, Walter GJ, Scheicher R, Bellutti F, Prchal-Murphy M, Tigan AS, Valent P, Heidel FH, Kubicek S, Scholl C, et al. Palbociclib treatment of FLT3-ITD+ 1 AML cells uncovers a kinase-dependent transcriptional regulation of FLT3 and PIM1 by CDK6. Blood. 2016; 127(23): 2890–2902. doi:10.1182/blood-2015-11.
- Kollmann K, Heller G, Schneckenleithner C, Warsch W, Scheicher R, Ott RG, Schäfer M, Fajmann S, Schlederer M, Schiefer AI, et al. A kinase-independent function of CDK6 links the cell cycle to tumor angiogenesis. Cancer Cell. 2013; 24(2): 167–181. doi:10.1016/j.ccr.2013.07.012.
- Kollmann K, Sexl V. CDK6 and p16 INK4A in lymphoid malignancies. Oncotarget. 2013;4(11):1858–1859. doi:10.18632/oncotarget.1541.
- Hydbring P, Malumbres M, Sicinski P. Non-canonical functions of cell cycle cyclins and cyclin-dependent kinases. Nat Rev Mol Cell Biol. 2016;17(5):280–292. doi:10.1038/nrm.2016.27.
- Bellutti F, Tigan A-S, Nebenfuehr S, Dolezal M, Zojer M, Grausenburger R, Hartenberger S, Kollmann S, Doma E, Prchal-Murphy M, et al. CDK6 Antagonizes p53-Induced Responses during Tumorigenesis. Cancer Discov. 2018; 8(7): 884–897. doi:10.1158/2159-8290.CD-17-0912.
- Li Y, Chitnis N, Nakagawa H, Kita Y, Natsugoe S, Yang Y, Li Z, Wasik M, Klein-Szanto AJP, Rustgi AK, et al. PRMT5 is required for lymphomagenesis triggered by multiple oncogenic drivers. Cancer Discov. 2015; 5(3): 288–303. doi:10.1158/2159-8290.CD-14-0625.
- Winter GE, Buckley DL, Paulk J, Roberts JM, Souza A, Dhe-Paganon S, Bradner JE. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science. 2015;348(6241):1376–1381. doi:10.1126/science.aab1433.
- Olson CM, Jiang B, Erb MA, Liang Y, Doctor ZM, Zhang Z, Zhang T, Kwiatkowski N, Boukhali M, Green JL, et al. Pharmacological perturbation of CDK9 using selective CDK9 inhibition or degradation. Nat Chem Biol. 2018; 14(2): 163–170. doi:10.1038/nchembio.2538.