
ABSTRACT
Protein misfolding and protein aggregation are linked to several diseases commonly called as proteinopathies, which include cancer. Understanding the mechanisms of proteostasis could provide newer strategies to combat proteinopathies. We have recently demonstrated a new mechanism where we found that TRIM16 (tripartite motif-containing protein 16) utilizing NRF2-p62 axis and autophagy streamlines the safe disposal of misfolded proteins to maintain protein homeostasis.
The protein misfolding and the protein aggregation is now extensively linked with cancer progression.Citation1-Citation4 Due to increased metabolic activities, the cancer cells are in enormous oxidative, and proteotoxic stress and both of these stresses can induce protein misfolding. The cancer cells have a heightened capacity to degrade the misfolded proteins and the protein aggregates utilizing the two protein quality control systems, the proteasome, and the autophagy, both of them are induced in cancer.Citation5,Citation6 The inhibition of both of the processes can lead to an increase in the cytotoxic misfolded proteins and other cellular waste ultimately killing the cancer cells.Citation7 Hence, this approach is being used in cancer therapeutics.Citation7 The misfolded proteins are much more toxic than the protein aggregates.Citation8 In some cases, the formation of proteins aggregates is helpful in cancer cell survival. A well-known case is of aggregation-prone mutant p53 proteins (TP53, best known as p53) which are shown to co-aggregate with wild-type p53 and the other tumor suppressor proteins resulting in loss of tumor suppressor function and increased cancer progression.Citation4 Given the role of misfolded proteins and protein aggregates in cancer and also other proteinopathies, it is important to understand the molecular mechanisms by which misfolded proteins are converted into aggregates and later degraded.
In this avenue, we recently showed that a TRIM (tripartite motif-containing) family protein called TRIM16 governs the process of protein aggregates formation and degradation by regulating two cell detoxifying systems, NRF2 (nuclear factor erythroid 2 like 2), and autophagy.Citation1 The TRIM16- or NRF2-depleted HeLa cells were defective in the oxidative/proteotoxic stress-induced biogenesis of protein aggregates (from misfolded proteins). We found that TRIM16 interacts with both NRF2 and p62/SQSTM1 (sequestosome-1) and utilizes multiple mechanisms to stabilize and activate NRF2 (). KEAP1 (Kelch-like ECH-associated protein 1) negatively regulates NRF2 by mediating its degradation; we found that TRIM16 displaces the KEAP1 from NRF2. In another mechanism, the TRIM16 induces K63-linked ubiquitination of NRF2 leading to its stabilization and activation (). We found that the SPRY (SPla and the RYanodine Receptor) domain of TRIM16 is essential for its most of the functions. The p62 and NRF2 interact at the SPRY domain of TRIM16. The SPRY domain is required for K63-linked ubiquitination, stabilization, and activity of NRF2. We showed that the activated NRF2 induces expression of ubiquitin and ubiquitin pathway proteins which along with p62 were required for the biogenesis of protein aggregates (). Taken together, we show that under oxidative and proteotoxic stress conditions, TRIM16 whose expression is itself controlled by oxidative stress and NRF2 activates NRF2 and downstream genes including p62 to formulate the conversion of misfolded proteins into protein aggregates (). Although the role of NRF2 in anti-oxidative stress response is well studied, our study for the first time connected the NRF2 directly with the stress-induced biogenesis of protein aggregates. This study for the first time also showed that NRF2 regulates expression of ubiquitin and several ubiquitin pathway genes including Ubb (Ubiquitin B), Uba-1 (ubiquitin-like modifier activating enzyme 1), Ube2h (ubiquitin-conjugating enzyme E2 H), Ube2n (ubiquitin-conjugating enzyme E2 N), and Ube2z (ubiquitin-conjugating enzyme E2 N).
Figure 1. TRIM16 controls proteostasis and cancer cell survival under stress conditions. The TRIM16 (tripartite motif-containing protein 16) complexes with NRF2 (nuclear factor erythroid 2 like 2) resulting in displacement and degradation of KEAP1 (Kelch-like ECH-associated protein 1). TRIM16 enhances K63-linked ubiquitination of NRF2 and stabilizes its activity leading to induction of program of genes required to convert stress-induced misfolded proteins into protein aggregates. This includes ubiquitin system proteins, p62 and TRIM16. TRIM16 also act as a scaffold protein to bring LC3B (microtubule associated protein 1 light chain 3 beta), ULK1 (Unc-51 like autophagy activating kinase 1), and ATG16L1 (Autophagy related 16 like 1) over the protein aggregates and hence mediate p62/LC3-dependent degradation of protein aggregates. By removing the misfolded proteins, TRIM16 maintains protein homeostasis of cells and survival of cancer cell in harsh oxidative or proteotoxic stress condition. The TRIM16 knock out (TRIM16−/-) tumors as compared to wild-type (TRIM16+/+) were regressed rapidly on exposure of oxidative/proteotoxic stress.
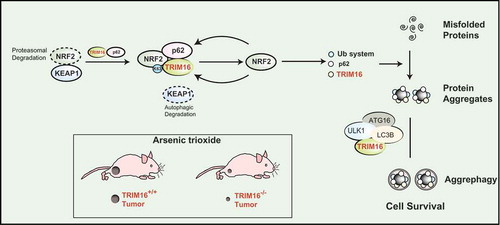
Selective autophagy, which is termed as aggrephagy plays a key role in the degradation of protein aggregates. TRIM family proteins are shown to orchestrate different kinds of selective autophagy.Citation9,Citation10 We found that TRIM16 not only directs the conversion of misfolded proteins into protein aggregates but also play an important role in the autophagic degradation of protein aggregates (). TRIM16 along with p62 is present over the protein aggregates and act as a scaffold protein to assemble autophagy machinery proteins LC3B (microtubule-associated protein 1 light chain 3 beta), ULK1 (Unc-51 like autophagy activating kinase 1), and ATG16L1 (Autophagy-related 16 like 1) over the protein aggregate for aggrephagy (). Thus, TRIM16 streamlines the process of safe clearance of misfolded proteins during oxidative and proteotoxic stress conditions and thus could protect from the cytotoxic effects of misfolded proteins. Indeed, we found that TRIM16 CRISPR knock out cells were highly sensitive to oxidative and proteotoxic stress-induced cell death.
A recent study suggests that enhanced proteasomal degradation of misfolded proteins via NRF2-TRIM11 axis helps cancer cells to survive in oxidative and proteotoxic stress conditions.Citation3 They found that increased capacity to degrade misfolded protein alleviate the oxidative stress associated with oncogenic growth and is required for both the initiation and maintenance of malignancy. In our study, we found that TRIM16-NRF2 axis and TRIM16-mediated autophagy is required for clearance of stress-induced misfolded proteins and protein aggregates. This enhanced clearance of both misfolded proteins and protein aggregates in wild-type cancer cells helps them to proliferate in xenograft mice model even in the presence of augmented oxidative stress whereas TRIM16 knock out tumor’s regressed rapidly when mice were exposed to oxidative stress (). Both of the above studies point towards an interesting conclusion that crippling the machinery required to resolve stress-induced misfolded protein can lead to cytotoxicity and tumor regression. Our work suggests that combinatorial targeting of NRF2/TRIM16 (and maybe other TRIM proteins) along with autophagy leading to increased cytotoxic misfolded proteins could be a novel therapeutic approach against cancer.
Acknowledgments
The work is supported by the Wellcome Trust/Department of Biotechnology (DBT) India Alliance (IA/I/15/2/502071) fellowship, ILS core funding (Department of Biotechnology, India), and Early Career Reward (SERB, ECR/2016/000478) to Santosh Chauhan. Subhash Mehto and Swati Chauhan are supported by fellowship from SERB (NPDF, PDF/2016/001697) and DST (SR/WOS-A/LS-9/2016).
Additional information
Funding
References
- Jena KK, Kolapalli SP, Mehto S, Nath P, Das B, Sahoo PK, Ahad A, Syed GH, Raghav SK, Senapati S, et al. TRIM16 controls assembly and degradation of protein aggregates by modulating the p62-NRF2 axis and autophagy. Embo J. 37;2018. doi:10.15252/embj.201798358.
- Bruning A, Juckstock J. Misfolded proteins: from little villains to little helpers in the fight against cancer. Front Oncol. 2015;5:47. doi:10.3389/fonc.2015.00047.
- Chen L, Brewer MD, Guo L, Wang R, Jiang P, Yang X. Enhanced degradation of misfolded proteins promotes tumorigenesis. Cell Rep. 2017;18:3143–3154. doi:10.1016/j.celrep.2017.03.010.
- Xu J, Reumers J, Couceiro JR, De Smet F, Gallardo R, Rudyak S, Cornelis A, Rozenski J, Zwolinska A, Marine J-C, et al. Gain of function of mutant p53 by coaggregation with multiple tumor suppressors. Nat Chem Biol. 2011;7:285–295. doi:10.1038/nchembio.546.
- Amaravadi R, Kimmelman AC, White E. Recent insights into the function of autophagy in cancer. Genes Dev. 2016;30:1913–1930. doi:10.1101/gad.287524.116.
- Tsvetkov P, Adler J, Myers N, Biran A, Reuven N, Shaul Y. Oncogenic addiction to high 26S proteasome level. Cell Death Dis. 2018;9:773. doi:10.1038/s41419-018-0806-4.
- Vogl, D. T., et al. Combined autophagy and proteasome inhibition: a phase 1 trial of hydroxychloroquine and bortezomib in patients with relapsed/refractory myeloma. Autophagy. 2014;10:1380–1390. doi:10.4161/auto.29264.
- Muchowski PJ. Protein misfolding, amyloid formation, and neurodegeneration: a critical role for molecular chaperones? Neuron. 35;2002:9–12.
- Chauhan S, Kumar S, Jain A, Ponpuak M, Mudd MH, Kimura T, Choi SW, Peters R, Mandell M, Bruun J-A, et al. TRIMs and galectins globally cooperate and TRIM16 and galectin-3 co-direct autophagy in endomembrane damage homeostasis. Dev Cell. 2016;39:13–27. doi:10.1016/j.devcel.2016.08.003.
- Mandell MA, Jain A, Arko-Mensah J, Chauhan S, Kimura T, Dinkins C, Silvestri G, Münch J, Kirchhoff F, Simonsen A, et al. TRIM proteins regulate autophagy and can target autophagic substrates by direct recognition. Dev Cell. 2014;30:394–409. doi:10.1016/j.devcel.2014.06.013.