
ABSTRACT
The mammalian target of rapamycin complex 1 (mTORC1) plays an important role in the development and progression of multiple cancers. Its activity is regulated by both growth factor and nutrient signals, and the branched-chain amino acid (BCAA) leucine plays an important and unique role in this process. Recently we found that cancers of the liver and multiple other tissues suppress the catabolism of BCAAs, thereby facilitating the chronic activation of mTORC1. Our results unveil how mTORC1’s nutrient-sensing arm can be manipulated by tumors, and suggest that restoring BCAA catabolism may help control mTORC1 activity in cancer cells.
Branched chain amino acids (BCAAs), consisting of leucine, isoleucine, and valine, are biologically important metabolites not only because they are incorporated into synthesized proteins, but also because they have additional physiological effects as free amino acids. Some of these effects, such as activation of the mammalian target of rapamycin complex 1 (mTORC1) pathway, are well characterized,Citation1 while others remain enigmatic.Citation2 As essential amino acids, BCAAs cannot be synthesized de novo, and thus their intracellular levels are regulated by the balance of intake from the diet, uptake into the cells, and catabolism. To quickly review the catabolic pathway, BCAAs are first converted to their corresponding α-ketoacids in a reversible reaction carried out by the branched-chain amino acid transaminase (BCAT) 1 or 2 (). In the process, the amino group from a BCAA is transferred to α-ketoglutarate to generate glutamate. Since this step is reversible, BCAAs and α-ketoglutarate can also be generated from glutamate and corresponding branched-chain α-ketoacids, depending on the physiological context. Next, the α-ketoacids are irreversibly decarboxylated and attached to a CoA group by the branched-chain ketoacid dehydrogenase (BCKDH) complex, which in many cases is considered a rate-limiting step.Citation3 After a series of additional irreversible enzymatic steps, the final products are largely succinyl-CoA and acetyl-CoA, which can be fed into the tricarboxylic acid (TCA) cycle.
Figure 1. The reprogramming of branched-chain amino acid (BCAA) catabolism supports tumor development and progression. Normal and proliferative regenerating tissues express basal levels of BCAA catabolic enzymes to maintain homeostatic levels of BCAAs in tissues. However, in cancers of the liver and multiple other tissues, the activity of distal BCAA catabolic enzymes is suppressed through the downregulation of expression and/or changes in post-translational modifications, e.g. hyperphosphorylation of the branched-chain ketoacid dehydrogenase (BCKDH) complex by the branched-chain ketoacid dehydrogenase kinase (BCKDK). This leads to the accumulation of BCAAs in pre-tumor and tumor tissues, and facilitates the chronic activation of the mammalian target of rapamycin (mTORC1). Although we did not observe any overt, beneficial effects of high branched chain amino acid transaminase (BCAT) 1 or 2 expression in liver cancers, the gray box highlights recently identified additional/alternative mechanisms by which enhanced BCAT1/2 activity can influence the development and progression of other cancers.
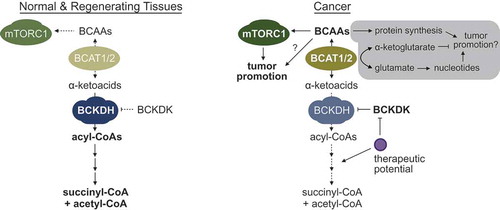
During our recent transcriptomic and metabolomic profiling of human hepatocellular carcinomas and animal models of liver cancer, we unexpectedly observed that the distal, irreversible steps of BCAA catabolism were potently and robustly suppressed in tumors, and that this correlated with an increase in tissue BCAA content.Citation4 Importantly, BCAA catabolic activity and BCAA content were unchanged in normal regenerating liver tissues, suggesting that the loss of BCAA oxidation is at least somewhat tumor-specific, and not simply needed for generic proliferation. By comprehensively analyzing the Cancer Genome Atlas (TCGA) datasets, we observed that suppression of BCAA catabolism was observed in a number of other cancers (most notably stomach, colorectal, and kidney), and independently predicted tumor aggressiveness and patient survival. In a series of follow-up experiments, we observed that changes in BCAA catabolism could potently regulate mTORC1 activity, and aid the development and progression of tumors. Notably, we also demonstrated that dietary BCAA intake significantly influences tissue BCAA content, and consequently, tumor development, progression, and survival.
While our manuscript was being reviewed, three other studies detailing how BCAAs are handled in certain cancers were published.Citation5-Citation7 Interestingly, while all groups observed and largely focused on an increased expression of BCAT1, they found opposing net fluxes and proposed alternate mechanisms. Specifically, some non-small cell lung cancers (NSCLCs) and chronic myeloid leukemias (CMLs) appear to enhance the conversion of BCAAs to α-ketoacids,Citation6,Citation7 while acute myeloid leukemias (AMLs) display a net production of BCAAs from α-ketoacids.Citation5 Why increased expression of the same enzyme leads to opposing net fluxes in the different cancers has not yet been clarified. It was suggested that NSCLCs enhance BCAA to α-ketoacid conversion to use the nitrogen in nucleotide synthesis, although knocking out BCAT1/2 did not affect cell proliferation in vitro, something that would conceivably be impacted by a lack of nucleotide precursors.Citation7 In the same study, pancreatic cancer was also examined, but we feel these results should be taken with caution given that the mouse model used has changes in key genes that are not consistent with human tumors, the TCGA pancreatic cancer dataset analyzed has a very low number of adjacent normal tissues (n = 4), and pancreatic cancers uniquely rely on macropinocytosis and lysosomal degradation of proteins rather than the uptake of free (and in this study, labeled) amino acids. In contrast to NSCLCs, enhanced BCAT1 expression in CMLs did not strongly affect nucleotide synthesis, but rather, helped keep α-ketoglutarate levels from going too high.Citation6 Although again, this is slightly curious given that there are 3 other transaminases in humans that can perform this job without utilizing precious (and frequently limited) essential amino acids such as the BCAAs. Finally, AMLs display enhanced production of BCAAs from α-ketoacids, which appears to stimulate mTORC1.Citation5
Given these findings, we also examined whether BCAT1 (or BCAT2) plays a significant role in the development of liver cancers. However, BCAT1/2 expression was not consistently different in human or animal liver cancers, and blocking BCAT1 activity actually enhanced the proliferation of the hepatoma cell line HepG2 by activating mTORC1. While tracing studies using C13/N15-labeled metabolites and additional in vivo models are needed to draw definitive conclusions, we currently have no evidence that enhanced BCAT1/2 expression in liver cancers play a critical role in tumor development or progression. In contrast, we largely focused on the distal, irreversible enzymatic steps, which were all robustly suppressed in tumors. Regardless of the flux through BCAT1/2, suppression of these distal enzymes spares both the BCAAs and α-ketoacids from oxidation to TCA intermediates, thereby increasing the intracellular pools of both metabolites, and facilitating the chronic activation of mTORC1. Indeed, restoring enzymatic activity via treatment with the branched-chain ketoacid dehydrogenase kinase (BCKDK) inhibitor BT2,Citation8 or re-expression of key catabolic enzymes potently suppresses mTORC1 activity and cancer cell proliferation. Thus far, we have seen a tight, direct correlation between sensitivity to BT2 and sensitivity to rapamycin across a large panel of cancer cell lines, further implicating an important mechanistic role for mTORC1.
It is helpful to point out a recent, elegant study by Zoltan Arany’s group that comprehensively characterized the whole-body metabolic fate of BCAAs in healthy, normal controls, and models of metabolic dysfunction.Citation9 Of particular interest, their results challenge a widely-accepted model that BCAAs and α-ketoacids need to be shuttled between multiple tissues for complete catabolism. In addition, they show unexpectedly that the normal pancreas consumes an uncharacteristically high level of BCAAs for both protein synthesis and oxidation. This raises caution about extrapolating results from pancreatic tissues and pancreatic cancer models, as they appear to be more of an outlier than the norm.
Overall, our studies not only address changes in BCAA catabolism in liver and other cancers, they reveal a unique mechanism whereby changes in cellular metabolism can promote carcinogenesis: through the stimulation of an established oncogenic signaling pathway. While mutations causing chronic stimulation of the growth factor arm of mTORC1 are broad and well established, our analyses found that tumors do not harbor a similar breadth of mutations stimulating the nutrient-sensing arm of mTORC1.Citation4 Thus, we believe tumors may instead manipulate the concentrations of specific metabolites to facilitate chronic activation. Ultimately, it will be helpful to comprehensively examine the consumption rate and metabolic fate of BCAA for all tissues/cancers by enzymatic assays and C13/N15-labeled metabolites, and examine the relative influence of the mechanisms involved, as they need not be mutually exclusive.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Shimobayashi M, Hall MN. Multiple amino acid sensing inputs to mTORC1. Cell Res. 2016;26:7–20. doi:10.1038/cr.2015.146.
- Taya Y, Ota Y, Wilkinson AC, Kanazawa A, Watarai H, Kasai M, Nakauchi H, Yamazaki S. Depleting dietary valine permits nonmyeloablative mouse hematopoietic stem cell transplantation. Science. 2016;354:1152–1155. doi:10.1126/science.aag3145.
- Shin AC, Fasshauer M, Filatova N, Grundell LA, Zielinski E, Zhou J-Y, Scherer T, Lindtner C, White PJ, Lapworth AL, et al. Brain insulin lowers circulating BCAA levels by inducing hepatic BCAA catabolism. Cell Metabolism. 2014;20:898–909. doi:10.1016/j.cmet.2014.09.003.
- Ericksen, RE, Lim, SL, McDonnell E, Shuen WH, Vadiveloo M, White PJ, Ding Z, Kwok R, Lee P, Radda GK, et al. Loss of BCAA catabolism during carcinogenesis enhances mTORC1 activity and promotes tumor development and progression. Cell Metabolism. 2019. doi:10.1016/j.cmet.2018.12.020.
- Hattori A, Tsunoda M, Konuma T, Kobayashi M, Nagy T, Glushka J, Tayyari F, McSkimming D, Kannan N, Tojo A, et al. Cancer progression by reprogrammed BCAA metabolism in myeloid leukaemia. Nature. 2017;545:500–504. doi:10.1038/nature22314.
- Raffel S, Falcone M, Kneisel N, Hansson J, Wang W, Lutz C, Bullinger L, Poschet G, Nonnenmacher Y, Barnert A, et al. BCAT1 restricts alphaKG levels in AML stem cells leading to IDHmut-like DNA hypermethylation. Nature. 2017;551:384–388. doi:10.1038/nature24294.
- Mayers JR, Torrence ME, Danai LV, Papagiannakopoulos T, Davidson SM, Bauer MR, Lau AN, Ji BW, Dixit PD, Hosios AM, et al. Tissue of origin dictates branched-chain amino acid metabolism in mutant Kras-driven cancers. Science. 2016;353:1161–1165. doi:10.1126/science.aaf5171.
- Tso SC, Gui W-J, Wu C-Y, Chuang JL, Qi X, Skvora KJ, Dork K, Wallace AL, Morlock LK, Lee BH, et al. Benzothiophene carboxylate derivatives as novel allosteric inhibitors of branched-chain alpha-ketoacid dehydrogenase kinase. J Biol Chem. 2014;289:20583–20593. doi:10.1074/jbc.M114.569251.
- Neinast MD, Jang C, Hui S, Murashige DS, Chu Q, Morscher RJ, Li X, Zhan L, White E, Anthony TG, et al. Quantitative analysis of the whole-body metabolic fate of branched-chain amino acids. Cell Metabolism. 2019;29:417–429 e414. doi:10.1016/j.cmet.2018.10.013.