
ABSTRACT
The binding of ligands to death receptors elicits distinct outcomes, such as apoptosis, inflammation and necroptosis, depending on the cellular context. We have recently described that the autophagic protein TP53INP2 favors apoptosis upon death receptor signaling and is a potential biomarker of responsiveness to TRAIL treatment.
Apoptosis, a highly regulated cellular suicide program, is common and conserved among metazoans. Various factors can induce apoptosis, among them death receptor ligands Fas ligand (FasL), tumor necrosis factor alpha (TNFα) and TNF related apoptosis inducing ligand (TRAIL) via death receptor oligomerization (Fas, TNF alpha receptor (TNFαR), death receptor 3 (DR3), death receptor 4 (DR4), and death receptor 5 (DR5)).Citation1 The triggering of death receptors by their ligands recruits numerous molecules to the forming intracellular complex, which in turn leads to two cellular outcomes, survival or death. Many studies have sought to identify components of these complexes that would favor the latter outcome, especially since the discovery of TRAIL ligand and its unique capacity to induce apoptosis in cancer cells without harming normal cells. Unfortunately, several clinical trials have failed due to the poor stability of soluble TRAIL protein and tumor resistance to TRAIL monotherapy. Consequently, there is a growing need for new therapeutic strategies.Citation2
We have recently reported a new regulator of death receptor signaling, namely the autophagy protein TP53INP2 (tumor protein p53-inducible nuclear protein 2).Citation3 We used several apoptotic inducers to identify the role of this protein in apoptosis and autophagy crosstalk. Our data showed that TP53INP2 sensitizes cancer cells to death receptor-induced apoptosis by acting as a scaffold for efficient ubiquitination of caspase-8 by TNF receptor-associated factor 6 (TRAF6). Furthermore, TP53INP2 and another autophagy protein, p62, aggregate ubiquitinated caspase-8 through their respective ubiquitin binding motifs to fully activate it.Citation3,Citation4 It is still unclear whether TP53INP2 is sufficient to aggregate ubiquitinated caspase-8. However, this function is in synergy with p62, as p62 knock-down only partially rescued death receptor-induced apoptosis. By screening several breast and liver cancer cell lines, we found a positive correlation between TP53INP2 protein levels and susceptibility of cancer cells to TRAIL. These findings thus identify TP53INP2 as a potential biomarker to distinguish patients who would benefit from TRAIL therapy.Citation3
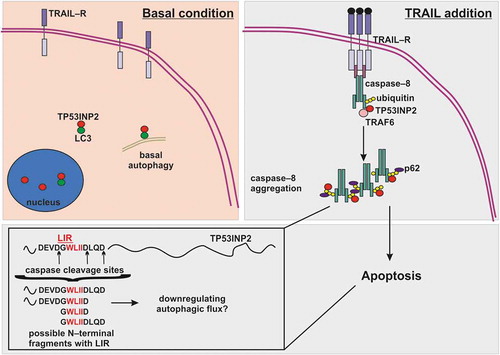
TP53INP2 is an intrinsically disordered protein involved in nuclear hormone receptor signaling, obesity, diabetes and autophagy.Citation5–Citation7 The function of TP53INP2 in death receptor signaling per se is independent of autophagy. However, the re-localization of this protein to the plasma membrane in response to TRAIL is likely to lower the amount of the protein available for autophagy, thus down-regulating autophagic flux and further favoring apoptosis. Indeed, a recent study showed that TP53INP2 regulates autophagy by acting as a scaffold for the lipidation of Microtubule-associated protein 1A/1B-light chain 3 (LC3) by Autophagy related 7 (ATG7).Citation8 The engagement of key autophagic proteins in apoptosis has the following advantages: (i) it facilitates the induction of apoptosis; and (ii) it sequesters autophagic proteins from their function in autophagy. Along the same lines, various studies have shown that targeting autophagy is a promising strategy to overcome TRAIL resistance in several types of cancer.Citation9 Furthermore, most autophagic proteins are cleaved after fulfilling their function of activating caspases in apoptosis, thereby impeding their role in autophagy and hence further favoring apoptosis. The same is true for TP53INP2 (). After activation of caspase-8, and probably caspase-3, TP53INP2 is cleaved. The caspase cleavage sites in TP53INP2 are around the LC3 interacting region (LIR) sequence at D33, D39 and D42 (). D33 and D42 are putative caspase-3 cleavage sites, while D39 is a putative caspase-8 cleavage site. The caspase that cleaves TP53INP2 first has not been identified to date; however, in the case of death receptor-induced apoptosis, we speculate that caspase-8 cleaves TP53INP2 at D39, thus cutting off the N-terminal part with the LIR sequence from the rest of the protein and probably abrogating its role in autophagy. Later, when caspase-3 is activated, additional cleavage(s) at D33 and D42 might take place, completely isolating the LIR sequence (W35-I38) from the rest of TP53INP2 (). It would be interesting to address whether cleavage at D39 by caspase-8 is enough to completely abrogate the function of TP53INP2 in autophagy, especially since it was shown that ATG7 binds to first 28 amino acids of TP53INP2. In addition, it is not clear if 1–39 amino acids long fragment of TP53INP2 is sufficient to facilitate LC3 lipidation.Citation8 Nevertheless, when caspase-3 is activated, the function of TP53INP2 in autophagy is abrogated. Moreover, the N-terminal caspase cleavage parts that still have the LIR sequence might further interfere with LC3 function in autophagy by omitting its binding to autophagy cargo receptor proteins like p62 or Neighbor of BRCA1 gene 1 (NBR1).Citation7
Figure 1. Role TP53INP2 in TRAIL-induced apoptosis. In basal condition TP53INP2 is localized in nucleus or in cytoplasm, where with LC3 has a function in basal autophagy. Upon TRAIL addition, TP53INP2 re-localizes to plasma membrane where acts as a scaffold for caspase-8 ubiquitination by TRAF6. Ubiquitinated caspase-8 is aggregated by p62 and TP53INP2 for its full activation, thus leading to apoptosis. Activated caspase-8 and effector caspases can cleave TP53INP2 at various aspartates (D) around the LIR sequence. The cleavage of TP53INP2 probably abrogates its function in autophagy and some of the possible N-terminal caspase cleavage fragments with LIR motif might further downregulate autophagy. LC3, Microtubule-associated protein 1A/1B-light chain 3; LIR, LC3 interacting region; TRAIL, Tumor necrosis factor (TNF) related apoptosis inducing ligand; TRAIL-R, TNF related apoptosis inducing ligand receptor; TRAF6, TNF receptor-associated factor 6, TP53INP2, tumor protein p53-inducible nuclear protein 2.
Autophagy signaling is almost invariably induced in response to TRAIL, and extensive studies have shown that autophagy is one of the mechanisms that causes resistance to TRAIL monotherapy.Citation9 Thus, inhibiting autophagy would have a synergistic effect when combined with TRAIL treatment. In fact, TP53INP2 sensitization to TRAIL might go in the same direction as TP53INP2 engagement in death receptor signaling and its cleavage by caspases most probably lowers autophagic flux. It would be interesting to examine whether TP53INP2 can also sensitize cancer cells that are intrinsically resistant to TRAIL-induced apoptosis, as occurs in mutant K-RAS (KRAS) expressing lung adenocarcinomas, in which TRAIL monotherapy could have pro-tumorigenic and pro-metastatic functions.Citation10 We speculate that, in this specific cellular context, TP53INP2 would sensitize cells to therapy combining autophagy inhibitors and TRAIL treatment. Further studies will reveal the specific cellular contexts in which TP53INP2 can be a potential biomarker for TRAIL monotherapy or combined therapies with TRAIL for personalized cancer treatments.
Hopefully, our recent study will shed some light on how to stratify cancer patients most likely to benefit from TRAIL treatment. Notably, autophagy inhibitors in combination with other standard chemotherapies like TRAIL are in various cancer clinical trials. Hence, we consider that TP53INP2 is a potential biomarker for TRAIL mono- or combined therapy.
Abbreviations
Disclosure of potential conflict of interests
No potential conflict of interests were disclosed.
Additional information
Funding
References
- Medema JP, Scaffidi C, Kischkel FC, Shevchenko A, Mann M, Krammer PH, Peter ME. FLICE is activated by association with the CD95 death-inducing signaling complex (DISC). Embo J. 1997;16:2794–2804. doi:10.1093/emboj/16.10.2794.
- de Miguel D, Lemke J, Anel A, Walczak H, Martinez-Lostao L. Onto better TRAILs for cancer treatment. Cell Death Differ. 2016;23:733–747. doi:10.1038/cdd.2015.174.
- Ivanova S, Polajnar M, Narbona-Perez AJ, Hernandez-Alvarez MI, Frager P, Slobodnyuk K, Plana N, Nebreda AR, Palacin M, Gomis RR, et al. Regulation of death receptor signaling by the autophagy protein TP53INP2. Embo J. 2019:pii: e99300. doi:10.15252/embj.201899300.
- Jin Z, Li Y, Pitti R, Lawrence D, Pham VC, Lill JR, Ashkenazi A. Cullin3-based polyubiquitination and p62-dependent aggregation of caspase-8 mediate extrinsic apoptosis signaling. Cell. 2009;137:721–735. doi:10.1016/j.cell.2009.03.015.
- Baumgartner BG, Orpinell M, Duran J, Ribas V, Burghardt HE, Bach D, Villar AV, Paz JC, Gonzalez M, Camps M, et al. Identification of a novel modulator of thyroid hormone receptor-mediated action. PLoS One. 2007;2:0001183. doi:10.1371/journal.pone.0001183.
- Mauvezin C, Orpinell M, Francis VA, Mansilla F, Duran J, Ribas V, Palacin M, Boya P, Teleman AA, Zorzano A. The nuclear cofactor DOR regulates autophagy in mammalian and Drosophila cells. EMBO Rep. 2010;11:37–44. doi:10.1038/embor.2009.242.
- Sala D, Ivanova S, Plana N, Ribas V, Duran J, Bach D, Turkseven S, Laville M, Vidal H, Karczewska-Kupczewska M, et al. Autophagy-regulating TP53INP2 mediates muscle wasting and is repressed in diabetes. J Clin Invest. 2014;124:1914–1927. doi:10.1172/JCI72327.
- You Z, Xu Y, Wan W, Zhou L, Li J, Zhou T, Shi Y, Liu W. TP53INP2 contributes to autophagosome formation by promoting LC3-ATG7 interaction. Autophagy. 2019;15:1–13. doi:10.1080/15548627.2019.1580510.
- Sharma A, Almasan A. Autophagy as mechanism of Apo2L/TRAIL resistance. Cancer Biol Ther. 2018;19(9):755–762. doi:10.1080/15384047.2018.1472191.
- von Karstedt S, Conti A, Nobis M, Montinaro A, Hartwig T, Lemke J, Legler K, Annewanter F, Campbell AD, Taraborrelli L, et al. Cancer cell-autonomous TRAIL-R signaling promotes KRAS-driven cancer progression, invasion, and metastasis. Cancer Cell. 2015;27:561–573. doi:10.1016/j.ccell.2015.02.014.