
ABSTRACT
While genomic instability and mitochondrial homeostasis are integral for cancer progression, how these two hallmarks interact remains poorly understood. Here, we reflect on the dialogue between chromatin-based genomic instability and impairment of mitochondrial function and depict the importance of this interaction in cancer progression to metastasis.
Maintaining genome stability is essential for organismal development and survival. Mammalian cells are endowed with multiple pathways that sense and repair DNA lesionsCitation1,Citation2. One deleterious type of lesions is DNA double-strand breaks (DSBs), whose repair involves phosphorylation of the histone variant H2AX on its Ser139 residue (called γH2AX)Citation3. Upon induction of DSBs, free DNA ends recruit the MRN (MRE11-RAD50-NBS1) complex, leading to the activation of the ATM (Ataxia telangiectasia mutated) kinase, which in turn phosphorylates H2AX. γH2AX initiates a signaling cascade, which includes the creation of binding sites for the MDC1 protein, that collectively function to activate DNA damage responses and promote repairCitation3. Additional players in this pathway include 53BP1, RNF8, UBC13, RAP80, and BRCA1, among many others. Compromising this pathway may lead to increased mutation loads, which can promote a broad range of human conditions including immune deficiency and cancer. In addition to the established evidence that suggests genomic instability plays a role in cancer cell aggressiveness, the importance of mitochondrial biogenesis in fostering cancer progression has also been explored in this regard. Indeed, impaired redox homeostasis resulting from defective mitochondrial respiration and genomic instability are tightly linked by the evidence that oxidative stress leads to elevated DNA lesions and chronic conditions that create an environment prone to the onset of cancer. Most importantly, mitochondrial respiration is an essential component of metabolic reprogramming during cancer progression to metastasis. However, there is a fundamental gap in our understanding of how genome repair controls mitochondrial metabolism in cancer cells.
Our previous work has provided the first comprehensive evidence of roles for histone H2AX and genomic instability in promoting spontaneous epithelial-to-mesenchymal transition (EMT) and colon cancer metastasisCitation4,Citation5. We have shown that this abnormal differentiation also occurs in epithelial breast cancer cell linesCitation6. However, when injected into mice, H2AX-deficient cells failed to establish enhanced metastasis at distant sites presumably because of failure to elicit metabolic reprogramming required to resume proliferation in a nutrients-deprived tumor microenvironment. In line with this observation, we found that H2ax depletion resulted in 60% increase in reactive oxygen species (ROS) levels in mouse embryonic fibroblasts (MEFs)Citation7. Mitochondrial ROS were highly elevated in H2ax-null cells, suggesting defective mitochondrial activity in H2ax mutants. These findings establish a physiological function of H2ax in mediating redox homeostasis. Which molecular mechanisms underlie H2ax deletion-induced oxidative stress? We conducted genome-wide differential gene expression analysis comparing H2ax-depleted and control mouse embryonic fibroblasts and found 21 ROS-responsive genes, of which Nfr2 appeared to be particularly prominent. Indeed, levels of Nrf2 and its transcriptional targets Nqo1 and Gclc were significantly reduced in the H2ax mutants. Analysis of the promoter activity of the antioxidant response element (ARE) binding site for Nrf2 revealed an impaired activity. When treated with buthionine sulfoximine (BSO), a specific inhibitor of glutamate-cysteine ligase (Gclc), one of the key enzymes in the biosynthesis of glutathione, the H2ax mutants failed to properly reactivate the antioxidant response genes. Ectopic expression of H2ax in knockout cells rescued the impaired ability of proper response to reactive oxygen species, while cells re-expressed with DNA repair-defective mutant form of H2ax (S139A) exhibited pattern of ROS response similar to H2ax-null cells Citation7. These findings corroborated our observations that oxidants, such as hydrogen peroxide (H2O2) and BSO, significantly reduced survival of H2ax-null cells. We concluded that H2ax depletion impairs the ability to elicit a proper response to oxidative stress via alteration of the Nrf2-regulated antioxidant pathwayCitation7. These findings indicated that histone H2ax is physiologically cytoprotective against oxidant-induced damage. Is H2ax a direct sensor of ROS or a protein of redox balance maintenance? Recently, an elegant report by Gruosso et al. demonstrated a role for ROS-mediated regulation of H2AX protein turnover in cancer therapyCitation8. The study has provided evidence that chronic oxidative stress mediates interaction of H2AX with an E3 ubiquitin ligase RNF168, which leads to enhanced H2AX protein degradation by the proteasome. As such, environments with chronic oxidative stress, aging or aggressive tumors, might repress H2AX expression. Our finding indicates that H2AX deletion per se led to excessive reactive oxygen species, thus reflecting the existence of a vicious cycle in which chronic oxidative stress leads to H2AX protein degradation, which in turn fosters a deleterious environment of enhanced oxidative damage. However, evidence is lacking on how H2AX loss-induced chromatin remodeling causes impairment of mitochondrial homeostasis.
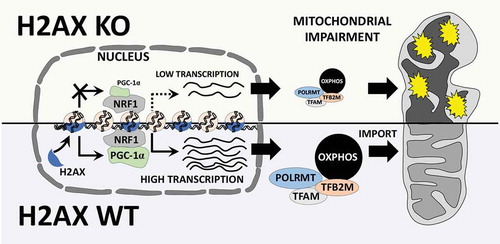
In our most recent paper, we demonstrated that increased genomic instability resulting from H2ax deletion leads to repression of Pgc-1α, a transcription co-activator essential for mitochondrial biogenesis and oxidative phosphorylation subunits (OXPHOS)Citation9 (). These data are highly substantiated by the evidence that transcriptional targets of Pgc-1α such as Tfam, Tfb2 m, Polrmt, and OXPHOS subunits were also reduced in H2AX mutants. Furthermore, H2ax-deficient MEFs exhibit impaired oxygen consumption rate, a characteristic which is reflective of impairment of mitochondrial respiration and consistent with the elevated ROS in H2ax-null cells. Similar observations were made in human colon cancer cell line HCT116 (data not shown). Other studies have reported that enhanced mitochondrial biogenesis through expression of Pgc-1α and elevated OXPHOS are required to promote anabolic metabolism, cell motility, invasiveness, cancer stemness, and metastatic colonization potential of cancer cellsCitation10. Together, we surmise that impaired OXPHOS resulting from DNA repair deficiency causes cancer cells to heavily rely on glycolysis, thus limiting the energy availability and synthesis of precursor metabolites for their proliferation in a nutrients-poor tumor environment. These new findings open up lines of investigation on the dialogue between chromatin-based genomic instability and mitochondrial homeostasis, and the relevance of this interaction in metabolic reprogramming of cancer cells. Therefore, an essential question arises: does chromatin have “a say” in the interplay between genomic instability and metabolism during cancer progression to metastasis?
Figure 1. Potential role for histone H2AX in mitochondrial homeostasis. H2AX is involved in fine control of mitochondrial biogenesis and respiration genes. Cells deficient for H2AX exhibit repression of the transcription co-activator PGC-1α (PPARG coactivator 1 alpha (PPARGC1A), best known as PGC-1α), and the subsequent diminution of PGC-1α transcription targets such as TFAM, TFB2 M, POLRMT, as well as reduction of oxidative phosphorylation subunits (OXPHOS). These alterations lead to deregulated import of proteins into the mitochondria, and enhanced repression of mitochondrial transcription, replication and repair processes. As a result, cells lacking histone H2AX exhibit accrued mitochondrial damages, as well as impairment of mitochondrial homeostasis.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Dr. Christophe Redon of the National Cancer Institute (NCI) for the figure design. The studies described in this commentary were supported by the National Institutes of Health (NIH) and USPHS grants MH18501 and DA000266. We thank Dr. William Bonner of the National Cancer Insitute, and Dr. Solomon H. Snyder of the Johns Hopkins University School of Medicine for their support and mentorship.
Additional information
Funding
References
- McKinnon PJ. Maintaining genome stability in the nervous system. Nat Neurosci. 2013;16:1–3. doi:10.1038/nn.3537.
- McKinnon PJ. DNA repair deficiency and neurological disease. Nat Rev Neurosci. 2009;10:100–112. doi:10.1038/nrn2559.
- Bonner WM, Redon CE, Dickey JS, Nakamura AJ, Sedelnikova OA, Solier S, Pommier Y. GammaH2AX and cancer. Nat Rev Cancer. 2008;8:957–967. doi:10.1038/nrc2523.
- Weyemi U, Redon CE, Choudhuri R, Aziz T, Maeda D, Boufraqech M, Parekh PR, Sethi TK, Kasoji M, Abrams N, et al. The histone variant H2A.X is a regulator of the epithelial-mesenchymal transition. Nat Commun. 2016;7:10711. doi:10.1038/ncomms10711.
- Weyemi U, Redon CE, Bonner WM. H2AX and EMT: deciphering beyond DNA repair. Cell Cycle. 2016;15:1305–1306. doi:10.1080/15384101.2016.1160659.
- Weyemi U, Redon CE, Sethi TK, Burrell AS, Jailwala P, Kasoji M, Abrams N, Merchant A, Bonner WM. Twist1 and Slug mediate H2AX-regulated epithelial-mesenchymal transition in breast cells. Cell Cycle. 2016;15:2398–2404. doi:10.1080/15384101.2016.1198864.
- Weyemi U, Paul BD, Snowman AM, Jailwala P, Nussenzweig A, Bonner WM, Snyder SH. Histone H2AX deficiency causes neurobehavioral deficits and impaired redox homeostasis. Nat Commun. 2018;9:1526. doi:10.1038/s41467-018-03948-9.
- Gruosso T, Mieulet V, Cardon M, Bourachot B, Kieffer Y, Devun F, Dubois T, Dutreix M, Vincent‐Salomon A, Miller KM, et al. Chronic oxidative stress promotes H2 AX protein degradation and enhances chemosensitivity in breast cancer patients. EMBO Mol Med. 2016;8:527–549. doi:10.15252/emmm.201505891.
- Weyemi U, Paul BD, Bhattacharya D, Malla AP, Boufraqech M, Harraz MM, Bonner WM, Snyder SH. Histone H2AX promotes neuronal health by controlling mitochondrial homeostasis. Proc Natl Acad Sci U S A. 2019;116:7471–7476. doi:10.1073/pnas.1820245116.
- LeBleu VS, O’Connell JT, Herrera KNG, Wikman H, Pantel K, Haigis MC, De Carvalho FM, Damascena A, Chinen LTD, Rocha RM, et al. PGC-1alpha mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat Cell Biol. 2014;16(992–1003):1001–1015. doi:10.1038/ncb3039.