
ABSTRACT
Renal medullary carcinoma (RMC) is a lethal disease that predominantly afflicts young individuals with sickle cell trait. Our recently reported molecular profiling of primary untreated RMC tissues elucidated distinct genomic and immune hallmarks of RMC, and identified MYC-induced replication stress as a targetable vulnerability for this disease.
Renal medullary carcinoma (RMC) is a lethal renal cell carcinoma (RCC) that predominantly afflicts young individuals of African descent with sickle cell trait or other sickle hemoglobinopathies.Citation1 All RMC tumors demonstrate loss by immunohistochemistry of the potent tumor suppressor SMARCB1 (also known as INI1, hSNF5 or BAF47), a subunit of the SWItch/Sucrose Non-Fermentable (SWI/SNF) chromatin remodeling complex.Citation2 The targeted therapies used for other RCCs are typically ineffective against RMC and the best available cytotoxic chemotherapies achieve a greater than 3 year overall survival in less than 5% of patients diagnosed with RMC.Citation1 There is clearly a need to develop new therapies informed by a deeper understanding of the biological pathways that drive RMC. We accordingly investigated the molecular landscape of RMC by performing comprehensive genomic and transcriptomic profiling of untreated primary RMC tisses.Citation3 To determine features unique to RMC, we compared our data to that of other closely related malignancies including collecting duct carcinoma (CDC), upper tract urothelial carcinoma (UTUC) and kidney malignant rhabdoid tumors (MRT). This integrative strategy produced new insights into the immune microenvironment of RMC, and identified a complex RMC genome harboring recurrent focal copy number alterations (CNAs). We further established a tumor-derived cell line and patient-derived xenograft (PDX) model of RMC, which allowed us to investigate a new treatment approach that exploits the therapeutic vulnerability to DNA damage repair (DDR) inhibition that we discovered characterizes this highly malignant tumor.Citation3
We found that the frequency of single-nucleotide variations (SNVs) in RMC is some of the lowest seen across all cancers. However, RMC is characterized by an abundance of larger structural alterations such as recurrent focal copy number alterations (often in chromosomal fragile sites), gain of chromosome 8q (where c-MYC is located), as well as deletions and inactivating translocations of the SMARCB1 gene.Citation3 Notably, we previously postulated that red blood cell sickling in the hypoxic renal medulla of individuals with sickle cell trait can induce such chromosomal structural alterations, particularly in known hotspots for genomic rearrangements.Citation2
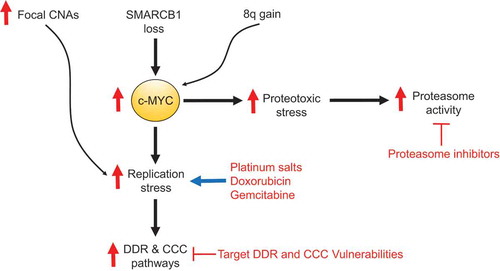
CDC arises from the same anatomical epicenter within the renal medulla and is morphologically very similar to RMC but expresses SMARCB1 by immunohistochemistry and is not associated with sickle hemoglobinopathies.Citation1–Citation3 UTUC originates anatomically close to the renal medulla and is treated with similar platinum-based cytotoxic chemotherapy regimens to those used for RMC.Citation1 MRT is negative for SMARCB1 by immunohistochemistry and can often arise from the kidney but morphologically appears like an undifferentiated rhabdoid malignancy, whereas RMC manifests as a high grade adenocarcinoma.Citation1,Citation4 Our transcriptomic analysis suggested that both RMC and CDC originate from the collecting ducts, in contrast to kidney MRTs.Citation3 Furthermore, the gene expression signature of RMC clearly distinguished this disease from UTUC and kidney MRT, whereas it shared similar core metabolic and hypoxia-associated gene expression patterns with CDC.Citation3 Conversely, pathways associated with MYC-induced replication stress were significantly upregulated in RMC compared with CDC tumors. Mechanistically, we demonstrated that SMARCB1 loss increases the binding of c-MYC to gene promoters that regulate DNA replication and cell cycle progression, and accelerates replication fork progression thus inducing replication stress and subsequent upregulation of DDR and cell cycle checkpoint (CCC) pathways.Citation3 Platinum salts, nucleoside analogs such as gemcitabine, and topoisomerase inhibitors such as doxorubicin can augment replication stress leading to cell deathCitation3 and this may explain why these chemotherapies are clinically effective against RMC.Citation1,Citation5 To prevent this mitotic catastrophe, RMC cells become dependent on their DDR and CCC pathways which can be targeted by drugs such as the poly ADP ribose polymerase (PARP) inhibitors nirapatib and olaparib or the WEE1 inhibitor adavosertib.Citation3 Furthermore, c-MYC activation by SMARCB1 loss increases proteotoxic stress rendering cells susceptible to perturbation of their proteostatic machinery by proteasome inhibitors such as bortezomib and ixazomib.Citation6,Citation7 Indeed, RMC cell lines and xenograft models are vulnerable to proteasome inhibitorsCitation6,Citation8 and there is evidence of clinical activity in patients with RMC that is more likely to be potent and durable if proteasome inhibitors are rationally combined with other therapies.Citation9 We have accordingly activated an ongoing phase II trial (NCT03587662 at clinicaltrials.gov) that targets both replication and proteotoxic stress by combining gemcitabine and doxorubicin with ixazomib ().
Figure 1. MYC-induced stress responses are renal medullary carcinoma (RMC) hallmarks. The loss of SMARCB1 and gain of 8q promote proteotoxic and replication stress responses mediated by c-MYC. The abundance of copy number alterations (CNAs) can be both a source and a consequence of replication stress which can be therapeutically targeted by agents that further induce replication stress including platinum salts, nucleoside analogs (such as gemcitabine) and topoisomerase inhibitors (such as doxorubicin). Replication stress may also be aggravated by the inhibition of DNA damage repair (DDR) pathways using drugs such as Poly (ADP-ribose) polymerase (PARP) inhibitors or the inhibition of cell cycle checkpoint (CCC) pathways using drugs such as the WEE1 inhibitor adavosertib. MYC-induced proteotoxic stress additionally confers a vulnerability to proteasome inhibitors such as ixazomib.
We noted that RMC does share some commonalities with urothelial carcinomas: 1) RMC profoundly upregulates the cancer-associated long non-coding RNA (lncRNA) urothelial cancer associated 1 (UCA1) to similar levels as those found in UTUC.Citation3 No other RCCs, including CDC, upregulate UCA1 and the biological role of this lncRNA in RMC remains to be elucidated. 2) RMC tumors often harbor NOTCH2 amplifications and concurrent deletions of NOTCH1 and NOTCH3,Citation3 a distinct pattern associated with increased aggressiveness in the basal subtype of bladder urothelial carcinoma.Citation10 Similarly to urothelial carcinoma, our results suggest that Notch pathway targeting in RMC should specifically focus on NOTCH2 inhibition.
Our immune profiling revealed RMC to be a highly inflamed tumor characterized by upregulation of the cyclic guanosine monophosphate–adenosine monophosphate synthase - stimulator of interferon genes (cGAS-STING) innate immune pathway, and an abundance of myeloid dendritic cells, neutrophils, and B lineage cells that distinguish RMC from kidney MRTs.Citation3 Although the expression of programmed death-ligand 1 (PD-L1) is heterogeneous, many immune checkpoints such as programmed cell death protein 1 (PD-1), cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), and lymphocyte-activation gene 3 (LAG3) are upregulated in RMC tumors.Citation3 We have accordingly activated a tissue-rich phase II trial (NCT03274258 at clinicaltrials.gov) to investigate the role of immune checkpoint inhibition in RMC. These ongoing clinical and co-clinical efforts will investigate why cGAS-STING is so upregulated, how best to harness immunotherapy, and what is the optimal strategy to target stress-induced vulnerabilities in RMC.
Author contributions
Manuscript preparation: P.M., C.L.W., G.G., N.M.T.
Disclosure statement
No potential conflicts of interest were disclosed.
Additional information
Funding
References
- Msaouel P, Hong AL, Mullen EA, Atkins MB, Walker CL, Lee CH, Carden MA, Genovese G, Linehan WM, Rao P, et al. Updated recommendations on the diagnosis, management, and clinical trial eligibility criteria for patients with renal medullary carcinoma. Clin Genitourin Cancer. 2019;17(1):1–3. doi:10.1016/j.clgc.2018.09.005.
- Msaouel P, NM T, CL W. A model linking sickle cell hemoglobinopathies and SMARCB1 Loss in Renal Medullary Carcinoma. Clin Cancer Res. 2018;24(9):2044–2049. doi:10.1158/1078-0432.CCR-17-3296.
- Msaouel P, Malouf GG, Su X, Yao H, Tripathi DN, Soeung M, Gao J, Rao P, Coarfa C, Creighton CJ, et al. Comprehensive molecular characterization identifies distinct genomic and immune hallmarks of Renal Medullary Carcinoma. Cancer Cell. 2020;37(5):720–734.e13. doi:10.1016/j.ccell.2020.04.002.
- Pawel BR. SMARCB1-deficient Tumors of childhood: a practical guide. Pediatr Dev Pathol. 2018;21(1):6–28. doi:10.1177/1093526617749671.
- Shah AY, Karam JA, Malouf GG, Rao P, Lim ZD, Jonasch E, Xiao L, Gao J, Vaishampayan UN, Heng DY, et al. Management and outcomes of patients with renal medullary carcinoma: a multicentre collaborative study. BJU Int. 2017;120(6):782–792. doi:10.1111/bju.13705.
- Carugo A, Minelli R, Sapio L, Soeung M, Carbone F, Robinson FS, Tepper J, Chen Z, Lovisa S, Svelto M, et al. p53 is a master regulator of proteostasis in SMARCB1-deficient malignant Rhabdoid tumors. Cancer Cell. 2019;35(2):204–220 e209. doi:10.1016/j.ccell.2019.01.006.
- Genovese G, Carugo A, Tepper J, Robinson FS, Li L, Svelto M, Nezi L, Corti D, Minelli R, Pettazzoni P, et al. Synthetic vulnerabilities of mesenchymal subpopulations in pancreatic cancer. Nature. 2017;542(7641):362–366. doi:10.1038/nature21064.
- Hong AL, Tseng YY, Wala JA, Kim WJ, Kynnap BD, Doshi MB, Kugener G, Sandoval GJ, Howard TP, Li J, et al. Renal medullary carcinomas depend upon SMARCB1 loss and are sensitive to proteasome inhibition. Elife. 2019;8. doi:(doi:10.7554/eLife.44161.
- Msaouel P, Carugo A, Genovese G. Targeting proteostasis and autophagy in SMARCB1-deficient malignancies: where next? Oncotarget. 2019;10(40):3979–3981. doi:10.18632/oncotarget.26970.
- Hayashi T, Gust KM, Wyatt AW, Goriki A, Jager W, Awrey S, Li N, Oo HZ, Altamirano-Dimas M, Buttyan R, et al. Not all notch is created equal: the Oncogenic role of NOTCH2 in Bladder Cancer and its implications for targeted therapy. Clin Cancer Res. 2016;22(12):2981–2992. doi:10.1158/1078-0432.CCR-15-2360.