
ABSTRACT
We recently developed a Brca1 coiled-coil mutant mouse model (Brca1CC). Brca1CC/CC results in embryonic lethality, with a fraction of mice reaching birth but with defects that parallel Fanconi anemia. Brca1CC/CC cells lacked Rad51 foci and were PARP inhibitor sensitive. Strikingly, inter-crossing with Brca1Δ11 generated Brca1CC/Δ11 mice that were developmentally normal.
When both parents carry a BRCA1 mutation, there is a 25% chance of progeny inheriting two mutant versions of the BRCA1 gene. Nevertheless, biallelic mutations are infrequently observed in patients due to the requirement for wild-type BRCA1 in embryo development.Citation1 In rare cases, individuals survive to birth and early adulthood, but present with developmental disorders similar to those observed in Fanconi anemia (FA) patients.Citation2–Citation5 Indeed, BRCA1 was also designated as the FANC-S complementation group. Similarly, homozygous Brca1 mutations in mice often induce embryonic lethality. In a recent study from our laboratory, we discovered that compound heterozygosity for particular combinations of Brca1 mutations can rescue developmental defects incurred by homozygous genotypes.Citation6
FA, embryonic lethality, and cancer predisposition are all associated with DNA repair defects. At double-stranded DNA breaks (DSB), BRCA1 functions to counteract 53BP1-RIF1 and promote DNA end resection. BRCA1 also directly interacts with PALB2 via their respective coiled-coil (CC) domains, forming a larger BRCA1-PALB2-BRCA2-RAD51 protein complex that is responsible for localizing the RAD51 recombinase to DSBs. RAD51 filaments mediate homology search and result in homologous recombination (HR) repair. While the role of BRCA1 in activating the DNA end resection step of HR is well established,Citation7,Citation8 the significance of BRCA1’s activity at the RAD51 loading step has been ambiguous.
Our study used Brca1 mutant mouse alleles to investigate the relative contribution of BRCA1 to DNA end resection initiation and the RAD51 loading steps of HR. To examine the role of BRCA1 in RAD51 loading, we developed a new Brca1 CC domain in-frame deletion mutant mouse model (Brca1CC), which generates a Brca1-CC protein. The BRCA1 CC domain functions to interact with PALB2; consequently, Brca1-CC did not interact with PALB2 and failed to promote loading of RAD51. Homozygosity for the Brca1CC allele results in late embryonic lethality, with few pups reaching birth. Brca1CC/CC mice exhibited FA-like defects and T-cell acute lymphoblastic leukemia (T-ALL) was responsible for shortened life-spans.
Because BRCA1-associated FA patients have an assortment of mutant allele combinations, we examined the effects of inter-crossing mice with different Brca1 mutations. The Brca1Δ11 allele produces a hypomorphic Brca1-Δ11 protein, while Brca1ΔC contains a truncation within the CC domain and fails to produce any detectable protein.Citation9 Homozygosity for Brca1ΔC and Brca1Δ11 both result in failed HR and embryonic lethality. Compound heterozygosity for Brca1ΔC/Δ11 and Brca1ΔC/CC allele combinations also resulted in embryonic lethality. However, inter-crossing mice with the Brca1CC and the Brca1Δ11 alleles, to our initial surprise, resulted in Brca1CC/Δ11 offspring that developed normally, had normal hematopoiesis, and were protected from cancer development.
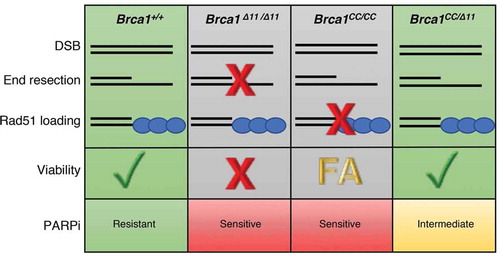
We hypothesized that proteins generated from Brca1CC and the Brca1Δ11 alleles may exhibit hypomorphic activity and perform compensatory functions within HR. Indeed, DNA end resection was severely compromised in Brca1Δ11/Δ11 mouse embryonic fibroblasts (MEFs), but Brca1CC/CC showed proficiency. Opposingly, Brca1CC/CC MEFs failed to promote Rad51 foci, while Brca1Δ11/Δ11 MEFs exhibited residual foci. In Brca1CC/Δ11 compound heterozygous MEFs, the protein products from each allele, Brca1-CC and Brca1-Δ11, were expressed, and MEFs showed end resection and RAD51 loading proficiency (). Consequently, Brca1CC/Δ11 MEFs were more PARPi resistant than either of the mutant homozygous genotypes. However, Brca1CC/Δ11 MEFs remained more sensitive to PARPi relative to Brca1+/+ MEFs, indicating that HR was only partially rescued and that combining mutant alleles does not provide the same level of HR proficiency as the Brca1 wild-type allele.
Figure 1. Link between homologous recombination functionality and development in Brca1 mutant mice. Wild-type Brca1 activity at DNA double strand breaks (DSB) supports the end resection and Rad51 loading steps of homologous recombination (HR), promoting viability and PARP inhibitor (PARPi) resistance. Brca1Δ11/Δ11 mouse embryonic fibroblasts (MEFs) produce the hypomorphic Brca1-Δ11 protein which does not promote end resection and results in dysfunctional HR, embryonic lethality, and sensitivity to PARPi. Brca1CC/CC MEFs express Brca1-CC and retain the ability to support end resection however fail to load Rad51 causing PARPi sensitivity, late embryonic lethality, and Fanconi anemia (FA)-like defects. In contrast, compound heterozygous Brca1CC/Δ11 mice exhibit resection and Rad51 loading due to the hypomorphic activities of Brca1-CC and Brca1-Δ11, respectively.
These results raised the question of whether human mutations can phenocopy mouse mutations. BRCA1 mutations in exon 11 are common due to the large size of the exon. We previously found that exon 11 mutations can express the BRCA1-Δ11q splice isoform, which has residual HR activity and is the homolog of the mouse Brca1-Δ11 protein. BRCA1 CC domain missense mutations are also observed in cancer patients and are known to disrupt the BRCA1-PALB2 interaction, including the L1407P and M1411T mutations. Similar to results with MEFs, human cells expressing CC domain missense mutations that disrupted the PALB2 interaction also showed severely reduced RAD51 loading, but end resection was relatively intact. Consequently, combining BRCA1-M1411T and BRCA1-Δ11q ectopic expression constructs also provided partial HR activity, indicating that functional complementation is possible in human cells.
In summary, the study by Nacson et al. emphasizes the dual activity of BRCA1 within HR. BRCA1 is not only important for DNA end resection, but also plays a critical role in directly recruiting RAD51 through the PALB2-BRCA2-RAD51 axis. Moreover, although HR activity is critical for development, recent publications indicate that partial HR activity is sufficient for mouse embryonic development.Citation9,Citation10 To date, patients with BRCA1 biallelic mutations have demonstrated severe abnormalities. However, research in mice now opens the possibility that BRCA1 genetic complementation might be possible in humans and could reduce the severity of FA.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Additional information
Funding
References
- Evers B, Jonkers J. Mouse models of BRCA1 and BRCA2 deficiency: past lessons, current understanding and future prospects. Oncogene. 2006;25(43):1–2. doi:10.1038/sj.onc.1209871.
- Domchek SM, Tang J, Stopfer J, Lilli DR, Hamel N, Tischkowitz M, Monteiro ANA, Messick TE, Powers J, Yonker A, et al. Biallelic deleterious BRCA1 mutations in a woman with early-onset ovarian cancer. Cancer Discov. 2013;3(4):399–405. doi:10.1158/2159-8290.CD-12-0421.
- Sawyer SL, Tian L, Kahkonen M, Schwartzentruber J, Kircher M, Majewski J, Dyment DA, Innes AM, Boycott KM, Moreau LA. Biallelic mutations in BRCA1 cause a new fanconi anemia subtype. Cancer Discov. 2015;5(2):135–142. doi:10.1158/2159-8290.CD-14-1156.
- Keupp K, Hampp S, Hubbel A, Maringa M, Kostezka S, Rhiem K, Waha A, Wappenschmidt B, Pujol R, Surrallés J, et al. Biallelic germline BRCA1 mutations in a patient with early onset breast cancer, mild Fanconi anemia-like phenotype, and no chromosome fragility. Molecular Genetics & Genomic Medicine. 2019;7(9):e863. doi:10.1002/mgg3.863.
- Seo A, Steinberg-Shemer O, Unal S, Casadei S, Walsh T, Gumruk F, Shalev S, Shimamura A, Akarsu NA, Tamary H, et al. Mechanism for survival of homozygous nonsense mutations in the tumor suppressor gene BRCA1. Proc Natl Acad Sci U S A. 2018;115(20):5241–5246. doi:10.1073/pnas.1801796115.
- Nacson J, Di Marcantonio D, Wang Y, Bernhardy AJ, Clausen E, Hua X, Cai KQ, Martinez E, Feng W, Callén E, et al. BRCA1 mutational complementation induces synthetic viability. Mol Cell. 2020;78(5):951–959.e6. doi:10.1016/j.molcel.2020.04.006.
- Bunting SF, Callén E, Wong N, Chen H-T, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao L; Bunting SF, Callen E, Wong N, Chen HT, Polato F, Gunn A, et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010;141(2):243–254. doi:10.1016/j.cell.2010.03.012.
- Bouwman P, Aly A, Escandell JM, Pieterse M, Bartkova J, van der Gulden H, Hiddingh S, Thanasoula M, Kulkarni A, Yang Q, et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat Struct Mol Biol. 2010;17(6):688–695. doi:10.1038/nsmb.1831.
- Nacson J, Krais JJ, Bernhardy AJ, Clausen E, Feng W, Wang Y, Nicolas E, Cai KQ, Tricarico R, Hua X, et al. BRCA1 mutation-specific responses to 53BP1 loss-induced homologous recombination and PARP inhibitor resistance. Cell Reports. 2018;24(13):3513–3527. e7. doi:10.1016/j.celrep.2018.08.086.
- Chen J, Li P, Song L, Bai L, Huen MSY, Liu Y, Lu L. 53BP1 loss rescues embryonic lethality but not genomic instability of BRCA1 total knockout mice. Cell Death Differ. 2020. doi:10.1038/s41418-020-0521-4.