
Abstract
Objective
To evaluate the value of nanopore targeted sequencing in diagnosing pneumonia pathogens.
Methods
This large-scale multicentre prospective study performed in 8 hospitals across China from April to October 2022. Hospitalised patients with a diagnosis of pneumonia at admission were included. Complete clinical data were collected, and bronchoalveolar lavage fluid were obtained from each patient. These samples underwent simultaneous testing using conventional microbial testing, metagenomic next-generation sequencing, and nanopore targeted sequencing.
Results
A total of 218 patients were included. Among the 168 cases of pulmonary infection, 246 strains of pathogens were confirmed. Nanopore targeted sequencing outperformed conventional microbial testing, identifying more pathogens with a sensitivity increase of 47.9% (77.2% vs. 29.3%). Metagenomic next-generation sequencing had a sensitivity of 82.9%. Total of 70.1% patients had consistent results in both metagenomic next-generation sequencing and nanopore targeted sequencing. Nanopore targeted sequencing exhibited significantly higher sensitivity in detecting Pneumocystis jiroveci, cytomegalovirus, Mycobacterium tuberculosis, Nontuberculous mycobacteria, Streptococcus pneumoniae, and Mycoplasma pneumoniae compared to conventional microbial testing. However, metagenomic next-generation sequencing demonstrated higher sensitivity than nanopore targeted sequencing for Aspergillus (88.5% vs. 53.8%). Regarding the detection of co-infections, nanopore targeted sequencing displayed significantly higher sensitivity than conventional microbial testing (76.7% vs. 28.7%) and was on par with metagenomic next-generation sequencing (76.7% vs. 82.9%).
Conclusion
Nanopore targeted sequencing performs equally well as metagenomic next-generation sequencing in bronchoalveolar lavage fluid for pathogen diagnosis in pneumonia, both methods showing higher sensitivity than conventional microbial testing. Nanopore targeted sequencing can be considered a reliable method for diagnosing pathogens in pneumonia.
Introduction
The key aspect of pneumonia diagnosis is identifying the pathogens involved. Culture-based diagnostic methods have drawbacks such as lengthy detection cycles, low sensitivity, and challenges in detecting atypical pathogens, viruses, and fastidious bacteria [Citation1]. On the other hand, polymerase chain reaction (PCR) based single-target or multi-target nucleic acid detection techniques offer rapid and sensitive pathogen detection but have limited coverage, capable of testing only 1–10 pathogens in a single assay [Citation2]. High-throughput, unbiased metagenomic next-generation sequencing (mNGS) can extensively cover pathogens with high sensitivity and a much broader range compared to multiplex PCR, making it valuable in identifying newly emerging pathogens [Citation3]. However, mNGS based on next-generation sequencing suffers from long detection cycles, high costs, complex bioinformatics processing, and difficulties in standardisation [Citation4]. These limitations have somewhat hindered the application and development of mNGS [Citation5–8].
To address these challenges, targeted amplification techniques offer advantages such as high specificity, low sample requirements, relatively simple laboratory procedures, and lower costs. They are not influenced by human genetic information, and both the experimental workflow and bioinformatics analysis are easily standardised. Nanopore sequencing technology is capable of performing bioinformatics analysis simultaneously with sequencing, which shortens the experimental time [Citation9,Citation10]. The Nanopore targeted sequencing (NTS) method combines nanopore sequencing technology with targeted amplification, such as bacterial 16S rRNA gene, Mycobacterium rpoB gene, and fungal internal transcribed spacer. NTS shows promise in overcoming the limitations of culture, PCR, and mNGS [Citation11].
In this study, we employed the faster and more cost-effective NTS technique for early pneumonia diagnosis, targeting the detection of the 188 most common respiratory pathogens (Supplementary 1 sTable S1). We conducted a prospective, multicentre study on pneumonia pathogen diagnosis in 8 hospitals in Zhejiang Province, China. Bronchoalveolar lavage fluid (BALF) samples were collected from patients clinically diagnosed with pneumonia and subjected to conventional microbial testing (CMTs), mNGS, and NTS. The aim of this study was to evaluate the diagnostic value of NTS in BALF from pneumonia patients by comparing it with multiple methods.
Materials and methods
Study population
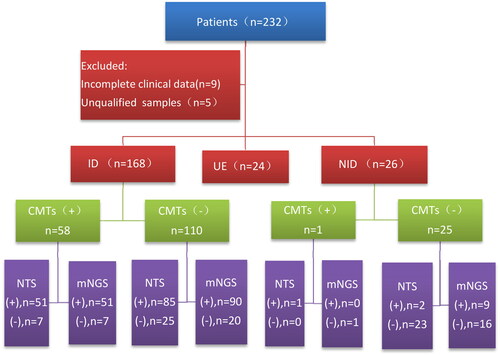
This study was conducted from April 2022 to October 2022 in 8 hospitals in Zhejiang, China. A total of 232 patients clinically diagnosed with pneumonia upon admission to the hospital were initially considered for inclusion. However, 14 cases were subsequently excluded, consisting of 9 cases with incomplete data and 5 cases with BALF samples that did not meet quality control requirements. Consequently, 218 patients were finally included in the study ().
Figure 1. Overall clinical diagnosis and pathogen detection outcomes of the recruited patients. A total of 232 patients underwent screening, with 218 of them being included in the study. The patients were categorised into three groups based on their conditions: the infectious disease group, the non-infectious disease group, and the unclear aetiology group. In order to assess the diagnostic efficacy of the detection methods, a comparative analysis of CMTs, mNGS, and NTS was conducted specifically for the infectious and non-infectious groups.
Inclusion and exclusion Criteria
The inclusion criteria were as follows: (1) Patients of any age or gender. (2) Pneumonia was diagnosed using the specific diagnostic criteria for community-acquired pneumonia and hospital-acquired pneumonia [Citation12,Citation13]. (3) Adequate BALF sample (>20 ml) was collected. (4) Patients able to read the informed consent form, understand and cooperate with the study protocol, and sign relevant documents.
The exclusion criteria were: (1) Sample issues: BALF samples that did not meet the quality control standards for mNGS or NTS. (2) Cases with incomplete data.
Data collection
Data were collected to provide information on the demographics, clinical characteristics, medical history, diagnostic and therapeutic interventions, radiological and laboratory findings, microbial testing results, pathologic data, and outcomes of the patients.
Sample and laboratory testing
All patients underwent bronchoscopy. BALF was collected at the site of the lesion selected based on pulmonary CT imaging, and the BALF was divided for further testing using CMTs, mNGS, and NTS.
Testing for CMTs
Routine microbial tests were conducted on BALF samples, including bacterial culture, mycobacterial culture, and fungal culture. Acid-fast staining for mycobacteria, fungal immunofluorescence staining, and tests for Aspergillus galactomannan antigen and Cryptococcus antigen were performed. Real-time PCR was used for the detection of Mycobacterium tuberculosis complex (MTB), Mycoplasma pneumoniae, Chlamydophila pneumoniae, cytomegalovirus (CMV), adenovirus, influenza A/B viruses, respiratory syncytial virus, human metapneumovirus, and other pathogens. Serum samples were also tested for (1,3)-β-D-glucan, galactomannan, and Cryptococcus antigen.
mNGS testing
BALF samples (>5 mL) were collected and immediately transported on dry ice to the Molecular Laboratory of the First Affiliated Hospital, Zhejiang University School of Medicine, for mNGS testing. The mNGS testing was performed within 8 h of sample collection. The detailed procedure for mNGS testing can be referenced from the study by Zhou et al. [Citation14].
NTS testing
Equivalent BALF samples (>5 mL) were collected and promptly transported on dry ice to Hangzhou Dian Medical Laboratory for NTS testing. For a detailed procedure of NTS testing, please refer to the attached document (Supplementary 2).
Criteria for positive mNGS results
The Results of Bacteria (excluding mycobacteria), viruses, parasites, fungi, MTB are according to the study by Langelier [Citation15–19]. Due to the balance between hospital and laboratory contamination [Citation20] and the low yield [Citation21], non-tuberculous mycobacteria (NTM) are considered positive when the mapped reads (genus or species level) rank among the top 10 in the bacterial list.
Criteria for positive NTS results
When the data size of a sample reaches 50 Mb and the number of detected sequences for the internal control (Lactococcus lactis) exceeds 100, the sample is deemed to have passed the quality control, and further analysis is conducted to determine positive species.
Alignment with a highly reliable local alignment search tool (BLAST)
The species information of each sequencing sequence is determined based on the species information of the database sequence to which it aligns. If over 80% of the database sequences in the BLAST results belong to a specific species, and the alignment coverage is not less than 85%, the species information is assigned to the sequencing sequence.
Pathogens not derived from environmental/reagent sources
The species results of the negative control sample are examined. If a species is detected in both the negative control and the test sample, but the number of reads for that species in the test sample exceeds three times that of the negative control, the species is retained. Otherwise, it is considered not to be a pathogenic microorganism and is discarded.
Pathogens not from cross-contamination in the same batch
The analysis focuses on the percentage of reads corresponding to a specific species within samples from the same batch. Any proportion below 0.5% is deemed to be the result of cross-contamination within that batch and cannot be interpreted as a positive finding.
Establishment of the final clinical diagnosis
The final clinical diagnosis of each participant enrolled in the study served as the benchmark for assessing the sensitivity and specificity of the detection methods. This diagnosis was established upon the patient’s discharge, following discussions between two respiratory physicians belonging to the medical team. In cases where a unanimous decision could not be reached, the research team organised a meeting with three experts to arrive at a conclusive diagnosis. The diagnosis took into account the patient’s clinical characteristics, findings from traditional pathogen detection, mNGS, NTS, pathology data, outcomes, and other relevant factors, facilitating a comprehensive assessment.
Irrespective of the coverage, the clinical significance of oral commensal bacteria requires meticulous evaluation based on clinical considerations. The determination of their clinical importance (Supplementary 3, sTable 2) necessitates thorough discussions and decision-making involving three experts from the research team. Unless such consensus is achieved, oral commensal bacteria cannot be classified as clinically significant microorganisms. These bacteria are commonly present in the human oropharynx, usually lacking clinical significance, and are easily detectable. Clinical practice, therefore, involves a careful analysis of these bacteria through comprehensive clinical assessment [Citation22].
Statistical analysis
Statistical analysis was conducted using SPSS 25.0 software (IBM Corporation, Chicago, Illinois, USA). Normally distributed continuous data were presented as mean ± standard deviation, while non-normally distributed continuous data were expressed as median and interquartile range (IQR, P25-P75). The clinical comprehensive diagnosis served as the reference standard, and to compare the diagnostic performance of CMTs, mNGS, and NTS, Pearson’s chi-square test or Fisher’s exact test was employed. All tests were two-tailed, and a p-value < .05 was deemed statistically significant.
Results
Basic characteristics of enrolled patients
A total of 218 patients from 8 hospitals were included in this study, comprising 131 males. The patients’ ages ranged from 16 to 90 years, with a mean age of 56 years. The most prevalent symptoms observed were cough, sputum production, and fever. Among the participants, 139 patients had one or more underlying diseases. The most common underlying disease category was respiratory system disorders (n = 80). After treatment, a significant proportion of patients (206/218; 94.5%) demonstrated clinical improvement or achieved complete cure. However, 10 patients did not exhibit any improvement in clinical symptoms, and unfortunately, 2 patients succumbed to the illness (Supplementary 4, sTable 3).
Clinical diagnosis of enrolled patients
Among the 218 patients, a total of 168 patients were categorised as having a pulmonary infection, with identifiable pathogens being the cause of their pneumonia. Among these 168 pulmonary infection patients, 115 were community acquired pneumonia, 7 were hospital acquired pneumonia, 46 were pneumonia of immunocompromised host. A group of 26 patients were confirmed to have non-infectious diseases. This group included various conditions, such as lung cancer (n = 4), pulmonary nodules (n = 6), organising pneumonia (n = 5), lymphoma with pulmonary involvement (n = 4), connective tissue disease-associated interstitial lung disease (n = 2), pulmonary haemorrhage (n = 2), radiation pneumonia (n = 1), eosinophilic granulomatosis with polyangiitis (n = 1), and allergic pneumonia (n = 1). There were 24 cases where the final diagnosis remained unclear, and the aetiology of their pneumonia could not be determined.
For the subsequent analysis of pathogen detection and diagnostic capabilities, data from 168 infectious patients and 26 non-infectious patients were included. However, the 24 cases classified as undetermined aetiology were excluded from the analysis.
Comparison of pathogenic diagnosis among CMTs, mNGS, and NTS
Among the 168 infectious patients, 63 showed positive results for CMTs, of which 40 cases completely matched the final pathogen diagnosis, 23 cases partially matched, and 105 cases did not match at all. Among the latter, 101 cases were negative for any pathogens, and 4 cases were detected with pathogens but were not considered pathogenic. As a result, the complete diagnosis rate was 23.8%, the partial diagnosis rate was 13.7%, and the undiagnosed rate was 54.1%. In the group of 26 non-infectious patients, 25 cases tested completely negative, while one case detected epstein-barr virus, which was not deemed the causative pathogen.
Out of the 168 infectious patients, 145 cases tested positive for mNGS, with 137 cases showing complete consistency with the final pathogen diagnosis, 8 cases partially consistent, and 23 cases completely inconsistent. Among the latter, 16 cases were negative for any pathogens, and 7 cases detected pathogens but were not considered the causative agents. Thus, the complete diagnosis rate was 81.5%, the partial diagnosis rate was 4.8%, and the undiagnosed rate was 13.7%. In the 26 non-infectious patients, 17 were completely negative, while 9 were found to have non-pathogenic results.
Regarding the NTS findings among the 168 infectious patients, 141 cases were detected, with 126 cases showing complete consistency with the final pathogen diagnosis, 15 cases partially consistent, and 27 cases completely inconsistent. Among the latter, 16 cases were negative for any pathogens, and 11 cases detected pathogens but were not considered pathogenic. Hence, the complete diagnosis rate was 75%, the partial diagnosis rate was 8.9%, and the undiagnosed rate was 16.1%. Among the 26 non-infectious patients, 23 were completely negative, while 3 detected pathogens were not identified as causative agents ().
Comparison of diagnostic performance between NTS and CMTs in pathogen detection
Among the 194 patients, consisting of 168 infectious cases and 26 non-infectious cases, 58 patients (29.9%) tested positive for both NTS and CMTs, while 44 patients (22.7%) were negative for both. A total of 86 patients (44.3%) showed positive results only for NTS, and 6 patients (3.1%) were solely positive for CMTs. Among the patients with both positive results, 34 out of 58 had completely matching outcomes, 4 cases were completely mismatched (all presenting mixed infections where both methods detected 1 pathogen but missed 1 pathogen), and the remaining 20 cases were classified as ‘partially matched’ (meaning that at least 1 pathogen was the same among the multiple microorganisms detected) (Supplementary 5, sFigure 1). Thus, the complete consistency rate between NTS and CMTs was 40.2% (78/194), and the partial consistency rate was 50.5% (98/194).
CMTs were unable to identify any effective pathogens in 130 out of 194 samples, while NTS successfully detected 86 out of 107 pathogens in the CMTs negative samples. Among the cases with unrecognised pathogens in NTS, CMTs identified 2 cases of actinomycetes, 2 cases of MTB, 1 case of Cryptococcus, and 1 case of adenovirus, totalling 6 cases.
In comparison to CMTs, NTS demonstrated superior pathogen detection across various categories, including bacteria (115 vs. 40), fungi (43 vs. 19), viruses (22 vs. 11), and other types of pathogens (10 vs. 2), which encompassed M. pneumoniae and Chlamydia Psittacosis (). Notably, CMTs failed to detect 10 pathogenic microorganisms that were successfully identified by NTS. These pathogens include S. pneumoniae, Moraxella catarrhalis, Stenotrophomonas maltophilia, anaerobes, C. Psittacosis, P. jiroveci, Penicillium marneffei, Rhizopus microsporis, Sedospora tip, and rhinovirus ().
Figure 2. Comparison of pathogen detection results using CMTs, mNGS, and NTS for various types of pathogens. The number of positive samples (y-axis) detected for bacterial, fungal, and viral groups (x-axis) is plotted using CMTs, mNGS, and NTS.
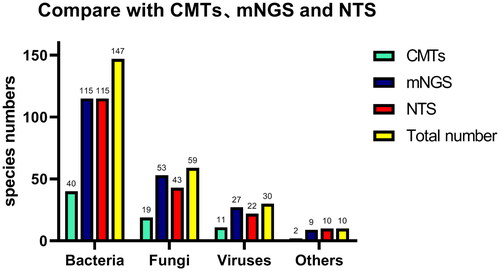
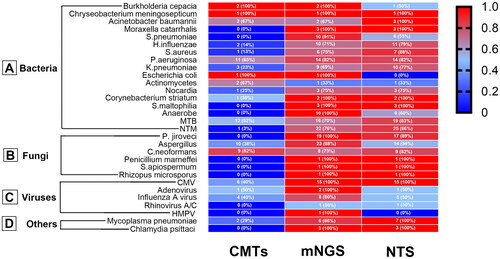
Figure 3. Heat map displaying the distribution of pathogens detected by CMTs, mNGS, and NTS. The positive rate of each detection method was calculated using clinically confirmed pathogens as the gold standard.
According to the final clinical diagnosis, a total of 246 responsible pathogens were detected in 168 infectious patients, with NTS identifying 190 strains (77.2%), whereas CMTs only identified 72 strains (29.3%). In the non-infectious group, NTS detected 5 non-responsible pathogens in 26 patients, while CMTs detected only one non-responsible pathogen (). A comparison between NTS and CMTs revealed a significant increase in pathogen detection sensitivity by 47.9% with NTS (77.2% vs. 29.3%; p < .01). The specificity of CMTs and NTS was 97.0% and 84.8%, respectively, with no statistically significant difference (p = .087). Both NTS and CMTs showed high positive predictive values of 97.4% and 98.6%, respectively, with no significant difference (p = .556). However, the negative predictive value of NTS was significantly higher than that of CMTs (33.3% vs. 15.5%; p < .01).
Table 1. Diagnostic performance of CMTs, mNGS, and NTS in identifying infectious disease.
Comparison of diagnostic performance between NTS and mNGS in pathogen detection
Among the 168 infectious patients, mNGS detected 204 strains of pathogens, while NTS identified 190 strains (). Both methods (mNGS vs. NTS) yielded comparable results concerning the number of detected bacteria (115 vs. 115), fungi (53 vs. 43), viruses (27 vs. 22), and other types of pathogens (9 vs. 10), p = .775. However, there were two responsible pathogens detected by mNGS that were not within the 188 pathogens of NTS, namely Aspergillus versicolour (n = 1) and Mycobacterium chimeranum (n = 1).
The majority of pathogens detected by NTS and mNGS are illustrated in . Among bacteria, the top three detected by both methods were MTB, NTM, and Pseudomonas aeruginosa (). In the case of fungi, the top three detected were Aspergillus, P. jiroveci, and Cryptococcus neoformans (). Regarding viruses, CMV and influenza A virus were the most frequently detected by both methods, while human metapneumovirus was exclusively detected by mNGS (). Lastly, among other types of pathogens, M. pneumoniae and Psittacosis were the most commonly detected ().
Out of the 194 cases, 122 (62.9%) tested positive for both NTS and mNGS, while 18 (9.3%) were negative in both methods. Among the double positive samples, 93 out of 122 patients’ results were completely matched, 4 were completely mismatched, and 25 were partially matched (Supplementary 6, sFigure 2). Thus, the complete consistency rate between NTS and mNGS was 57.2% (111/194), and the partial consistency rate was 70.1% (136/194). Among the four patients with completely inconsistent results between the two test methods in the double positive samples, the different pathogens detected by each method held clinical significance. Additionally, there were 22 samples (11.3%) that tested only positive for NTS, and 32 samples (16.5%) that tested only positive for mNGS. Using mNGS, 39 strains of microorganisms were identified in 32 patients who had undetectable effective pathogens in NTS. These included Aspergillus spp. (n = 7), MTB (n = 4), Prevotella niger (n = 3), NTM (n = 2), human metapneumovirus (n = 1), adenovirus (n = 1), P. jiroveci (n = 1), influenza A (n = 1), Mycobacterium abscesses (n = 1), and actinomycetes (n = 1) as the identified pathogens. On the other hand, NTS detected 27 strains of microorganisms in 22 cases that were not recognised by mNGS. These pathogens included MTB (n = 4), Aspergillus (n = 3), NTM (n = 2), M. pneumoniae (n = 1), Nocardia abscesses (n = 1), Actinomycetes cariae (n = 1), Cryptococcus neoformans (n = 1), and influenza A virus (n = 1).
Out of the 246 pathogenic pathogens identified in 168 patients with infectious diseases, mNGS was able to diagnose 204, while NTS could diagnose 190, showing equivalent sensitivity for both methods (82.9% vs. 77.2%; p = .114). Among the 26 non-infectious cases, NTS detected 5 non-responsible pathogens, whereas mNGS detected 15 non-responsible pathogens (). The specificity of NTS was higher than that of mNGS (84.8% vs. 54.5%; p < .01). The negative predictive values of mNGS and NTS were comparable, at 30.0% and 33.3% respectively (p = .672). However, the positive predictive value of NTS was higher than that of mNGS (97.4% vs. 93.2%; p < .05).
Comparison of detection performance between NTS and CMTs in specific pathogens
Regarding the detection of P. jiroveci, the sensitivity of NTS was significantly higher than that of hexamine silver staining in this study (p < .05). The detection sensitivity for Aspergillus, Cryptococcus, and influenza viruses was found to be similar to that of CMTs. However, for the detection sensitivity of CMV, NTM, MTB, and S. pneumoniae, NTS exhibited significantly higher sensitivity compared to CMTs ().
Table 2. Comparison of the sensitivity of CMTs, mNGS, and NTS to 9 pathogens.
Comparison of detection performance between NTS and mNGS in specific pathogens
NTS and mNGS exhibit similar sensitivity in detecting P. jiroveci, Cryptococcus, influenza, CMV, NTM, MTB, M. pneumoniae, and S. pneumoniae. However, in the case of Aspergillus, mNGS demonstrated significantly higher sensitivity than NTS (88.5% vs. 53.8%, p < .01) ().
Comparison in diagnosis of mixed infections
Among the 56 patients diagnosed with a mixed infection, a total of 129 pathogens were identified, with 37 detected by CMTs, 107 by mNGS, and 99 by NTS (Supplementary 7, sFigure 3). The sensitivity of NTS was significantly higher than that of CMTs (76.7% vs. 28.7%, p < .01), whereas the sensitivity of NTS and mNGS was found to be equivalent (76.7% vs. 82.9%, p = .214). The top 12 pathogens detected in patients with mixed infection pneumonia, Aspergillus was the most common pathogen (14/56, 25.0%), followed by P. jiroveci (13/56, 23.2%), and CMV (12/56, 21.4%) (Supplementary 8, sFigure 4).
Discussion
Lower respiratory tract infection remains one of the world’s deadliest infectious diseases [Citation23]. Delayed aetiological diagnosis can result in inappropriate empirical broad-spectrum antibiotic treatment, leading to poor treatment outcomes, prolonged hospital stays, and increased medical expenses [Citation24]. Therefore, early microbial identification is critical for the timely diagnosis and treatment of pneumonia patients.
Traditional microbiological culture methods have drawbacks [Citation25], using traditional pathogen detection methods, only 38% of pneumonia patients have their pathogens identified [Citation26]. Multiplex PCR analysis has proven to be valuable in enhancing the detection of pathogens in the lower respiratory tract [Citation27]. These products come in single-target formats (e.g. COVID-19) or multi-target formats (e.g. FilmArray, which covers the seven most common pathogens of community-acquired pneumonia). However, even multi-target PCR is limited to a few key pathogens. On the other hand, mNGS offers objective, accurate, comprehensive, and rapid detection of pathogenic microorganisms in samples. It does not rely on clinical prediction and is not restricted to rare or uncommon pathogens, making it capable of even detecting new pathogens.
The core technology for high-throughput sequencing of metagenomes currently relies on second-generation sequencing. Due to its long development history, high accuracy of base results, and relatively low cost, second-generation sequencing is more widely used at present. However, its reading length (50–100 bp) poses challenges in data analysis, affecting the efficiency and accuracy of splicing and assembly, making it difficult to distinguish similar pathogens, and also hindering the prediction of drug resistance in strains. Moreover, its operation is complex, sequencing time is long, and the cost is high. It requires accumulating a certain sample size for batch testing. These limitations prevent second-generation sequencing from meeting the clinical requirements for rapid and accurate pathogen diagnosis, pathogen identification, and integrated reporting of drug resistance results in respiratory infectious diseases. Consequently, its future promotion and widespread use are challenging.
On the other hand, the third-generation sequencing technology of nanopores abandons the fluorescence signal-based detection method used in second-generation sequencing. Instead, it directly reads base information by detecting the current changes that occur when nucleic acid molecules pass through the nanopores [Citation28]. This technology offers fast sequencing speed (sequencing speed > 400 bp/s) and long read lengths (average read length > 1000 bp), which effectively improves the efficiency and accuracy of splicing and assembly, enabling more precise detection of various infectious pathogens [Citation28]. NTS based on nanopore sequencing technology, combined with ultra-multiple PCR amplification and nanopore high-throughput sequencing, allows direct detection of hundreds of known pathogenic microorganisms in clinical samples [Citation29]. It has garnered increasing attention in the field of infectious pathogen diagnosis. Compared to metagenomic sequencing methods, NTS boasts advantages such as immunity to interference from human host content, a clear pathogen spectrum range, short sequencing time, and low detection cost.
In our study, we conducted a systematic comparison of NTS, mNGS, and CMTs in a paired manner and observed several advantages of NTS over the other methods. In comparison to CMTs, NTS demonstrated a remarkable 47.9% increase in sensitivity, a finding consistent with previous studies. For instance, Fu et al. [Citation5] reported that NTS detected pathogens in 88.7% of patient specimens, while culture methods only detected 37.8%. Several reasons contribute to the significant difference in sensitivity between CMTs and NTS. In this study, NTS showed significantly higher sensitivity in detecting six pathogens (P. jiroveci, CMV, MTB, NTM, S. pneumoniae, M. pneumoniae) compared to CMTs. The detection efficiency of P. jiroveci and NTM was partly affected in this study due to the absence of routine PCR detection for P. jiroveci and NTM. This indicates that the sensitivity of immunofluorescence staining for P. jiroveci and the detection efficiency of acid-fast staining and mycobacterium culture in NTM are not as high as that of NTS. Additionally, this study routinely conducted GeneXpert detection for MTB, but the detection sensitivity was lower than that of NTS, in line with previous research findings [Citation30]. This suggests that the NTS and MTB GeneXpert complement each other in diagnosing tuberculosis. Furthermore, the culture conditions for S. pneumoniae are harsh and may be affected by the use of empirical antibiotics, whereas NTS is less influenced by antibiotics.
Regarding sensitivity, NTS and mNGS exhibited comparable results, but NTS showed higher specificity compared to mNGS. The unbiased detection technology of mNGS allows for simultaneous detection of multiple pathogens, which provides advantages in diagnosing co-infections [Citation31].
The cost and time of mNGS and NTS are really quite important. We did not consider them as observed indicator of our research, which was an important limitation of our research. In fact, the experimental process and data analysis of mNGS takes approximately 24 h, from specimen collection to final report taking 48–72 h. The experimental process and data analysis of NTS takes approximately 8 h, and from specimen collection to final report taking about 24 h. Notably, the cost of an mNGS test in China was about ¥3500 (∼$500). The cost of our NTS was about ¥1000 (∼$150), only about one-third of mNGS. Obviously, NTS is much more affordable and timely.
Conclusion
The detection of pathogenic microorganisms in the BALF of pneumonia patients showed comparable performance between NTS and mNGS tests, with both significantly outperforming CMTs. Therefore, NTS can be considered a reliable method for diagnosing pulmonary infection pathogens.
Authors contributions
Hua Zhou, Yunsong Yu, Xi Li, Yan Chen, Huiming Guo, Yake Yao, Jianying Zhou and Qinqing Lin contributed in the design, data collection and analysis, and writing of the manuscript. Shanshan Zhang, Xiaolong Ma, Wenyu Chen, Chuhui Ru, Limin Wang, Bin Wang, Qiang Ma, Junfei Zhu, Xuemei Lin, Qi Chen, Hui Lou, Qi Chen, Junjun Chen and Zhu Zeng contributed to the data collection, data analysis, diagnosis and treatment of patients. Hua Zhou, Xi Li and Yunsong Yu contributed as data manager, building of the database, data analysis. Hua Zhou, Yunsong YU, Yake Yao, Xi Li and Yan Chen contributed to the data results interpretation and revision of the manuscript.
Ethical approval
This study has received ethical approval from the Ethics Committee of the First Affiliated Hospital, Zhejiang University School of Medicine (Hangzhou, Zhejiang, China) with the approval number IIT20220205B-R1. Informed consent forms were duly signed by all participating patients or their respective family members.
Supplemental Material
Download MS Word (66.6 KB)Supplemental Material
Download MS Word (98.6 KB)Supplemental Material
Download MS Word (38.8 KB)Supplemental Material
Download MS Word (36.9 KB)Supplemental Material
Download MS Word (17.6 KB)Supplemental Material
Download MS Word (17.4 KB)Supplemental Material
Download MS Word (26 KB)Supplemental Material
Download MS Word (23.3 KB)Disclosure statement
Qi Chen and Huiming Guo are employees of Zhejiang Digena Diagnosis Technology CO., LTD. The rest of the authors declared no conflict of interest.
Additional information
Funding
References
- Rajapaksha P, Elbourne A, Gangadoo S, et al. A review of methods for the detection of pathogenic microorganisms. Analyst. 2019;144(2):396–411. doi: 10.1039/c8an01488d.
- Simner PJ, Miller HB, Breitwieser FP, et al. Development and optimization of metagenomic next-generation sequencing methods for cerebrospinal fluid diagnostics. J Clin Microbiol. 2018;56(9):e00472–e00518. doi: 10.1128/jcm.00472-18.
- Gu W, Miller S, Chiu CY. Clinical metagenomic next-generation sequencing for pathogen detection. Annu Rev Pathol. 2019;14(1):319–338. doi: 10.1146/annurev-pathmechdis-012418-012751.
- Goodwin S, McPherson JD, McCombie WR. Coming of age: ten years of next-generation sequencing technologies. Nat Rev Genet. 2016;17(6):333–351. doi: 10.1038/nrg.2016.49.
- Fu Y, Chen Q, Xiong M, et al. Clinical performance of nanopore targeted sequencing for diagnosing infectious diseases. Microbiol Spectr. 2022;10(2):e0027022. doi: 10.1128/spectrum.00270-22.
- Charalampous T, Kay GL, Richardson H, et al. Nanopore metagenomics enables rapid clinical diagnosis of bacterial lower respiratory infection. Nat Biotechnol. 2019;37(7):783–792. doi: 10.1038/s41587-019-0156-5.
- Cheng J, Hu H, Kang Y, et al. Identification of pathogens in culture-negative infective endocarditis cases by metagenomic analysis. Ann Clin Microbiol Antimicrob. 2018;17(1):43. doi: 10.1186/s12941-018-0294-5.
- Schmidt K, Mwaigwisya S, Crossman LC, et al. Identification of bacterial pathogens and antimicrobial resistance directly from clinical urines by nanopore-based metagenomic sequencing. J Antimicrob Chemother. 2017;72(1):104–114. doi: 10.1093/jac/dkw397.
- Au CH, Ho DN, Ip BBK, et al. Rapid detection of chromosomal translocation and precise breakpoint characterization in acute myeloid leukemia by nanopore long-read sequencing. Cancer Genet. 2019;239:22–25. doi: 10.1016/j.cancergen.2019.08.005.
- Yu MC, Hung CS, Huang CK, et al. Integrative utility of long read sequencing-based whole genome analysis and phenotypic assay on differentiating isoniazid-resistant signature of Mycobacterium tuberculosis. J Biomed Sci. 2021;28(1):86. doi: 10.1186/s12929-021-00783-x.
- Jain M, Koren S, Miga KH, et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat Biotechnol. 2018;36(4):338–345. doi: 10.1038/nbt.4060.
- Cao B, Huang Y, She DY, et al. Diagnosis and treatment of community-acquired pneumonia in adults: 2016 clinical practice guidelines by the Chinese thoracic society, Chinese medical association. Clin Respir J. 2018;12(4):1320–1360. doi: 10.1111/crj.12674.
- Torres A, Niederman MS, Chastre J, et al. International ERS/ESICM/ESCMID/ALAT guidelines for the management of hospital-acquired pneumonia and ventilator-associated pneumonia: guidelines for the management of Hospital-Acquired Pneumonia (HAP)/Ventilator-Associated Pneumonia (VAP) of the European Respiratory Society (ERS), European Society of Intensive Care Medicine (ESICM), European Society of Clinical Microbiology and Infectious Diseases (ESCMID) and asociación latinoamericana del tórax (ALAT). Eur Respir J. 2017;50(3):1700582. doi: 10.1183/13993003.00582-2017.
- Zhou H, Larkin PMK, Zhao D, et al. Clinical impact of metagenomic next-generation sequencing of bronchoalveolar lavage in the diagnosis and management of pneumonia: a multicenter prospective observational study. J Mol Diagn. 2021;23(10):1259–1268. doi: 10.1016/j.jmoldx.2021.06.007.
- Langelier C, Zinter MS, Kalantar K, et al. Metagenomic sequencing detects respiratory pathogens in hematopoietic cellular transplant patients. Am J Respir Crit Care Med. 2018;197(4):524–528. doi: 10.1164/rccm.201706-1097LE.
- Bittinger K, Charlson ES, Loy E, et al. Improved characterization of medically relevant fungi in the human respiratory tract using next-generation sequencing. Genome Biol. 2014;15(10):487. doi: 10.1186/s13059-014-0487-y.
- Schlaberg R, Chiu CY, Miller S, et al. Validation of metagenomic next-generation sequencing tests for universal pathogen detection. Arch Pathol Lab Med. 2017;141(6):776–786. doi: 10.5858/arpa.2016-0539-RA.
- Doughty EL, Sergeant MJ, Adetifa I, et al. Culture-independent detection and characterisation of Mycobacterium tuberculosis and M. africanum in sputum samples using shotgun metagenomics on a benchtop sequencer. PeerJ. 2014;2:e585. doi: 10.7717/peerj.585.
- Simner PJ, Miller S, Carroll KC. Understanding the promises and hurdles of metagenomic next-generation sequencing as a diagnostic tool for infectious diseases. Clin Infect Dis. 2018;66(5):778–788. doi: 10.1093/cid/cix881.
- van Ingen J, Kohl TA, Kranzer K, et al. Global outbreak of severe Mycobacterium chimaera disease after cardiac surgery: a molecular epidemiological study. Lancet Infect Dis. 2017;17(10):1033–1041. doi: 10.1016/s1473-3099(17)30324-9.
- Özçolpan OO, Sürücüoğlu S, Özkütük N, et al. Distribution of nontuberculous mycobacteria isolated from clinical specimens and identified with DNA sequence analysis. Mikrobiyol Bul. 2015;49(4):484–493. doi: 10.5578/mb.9698.
- Graf EH, Simmon KE, Tardif KD, et al. Unbiased detection of respiratory viruses by use of RNA sequencing-based metagenomics: a systematic comparison to a commercial PCR panel. J Clin Microbiol. 2016;54(4):1000–1007. doi: 10.1128/jcm.03060-15.
- Hill UG, Floto RA, Haworth CS. Non-tuberculous mycobacteria in cystic fibrosis. J R Soc Med. 2012;105(Suppl 2):S14–S18. doi: 10.1258/jrsm.2012.12s003.
- Webb BJ, Sorensen J, Jephson A, et al. Broad-spectrum antibiotic use and poor outcomes in community-onset pneumonia: a cohort study. Eur Respir J. 2019;54(1):1900057. doi: 10.1183/13993003.00057-2019.
- Holter JC, Müller F, Bjørang O, et al. Etiology of community-acquired pneumonia and diagnostic yields of microbiological methods: a 3-year prospective study in Norway. BMC Infect Dis. 2015;15(1):64. doi: 10.1186/s12879-015-0803-5.
- Jain S, Self WH, Wunderink RG, et al. Community-acquired pneumonia requiring hospitalization among U.S. Adults. N Engl J Med. 2015;373(5):415–427. doi: 10.1056/NEJMoa1500245.
- Gadsby NJ, Russell CD, McHugh MP, et al. Comprehensive molecular testing for respiratory pathogens in community-acquired pneumonia. Clin Infect Dis. 2016;62(7):817–823. doi: 10.1093/cid/civ1214.
- Ciuffreda L, Rodríguez-Pérez H, Flores C. Nanopore sequencing and its application to the study of microbial communities. Comput Struct Biotechnol J. 2021;19:1497–1511. doi: 10.1016/j.csbj.2021.02.020.
- Zhang Y, Lu X, Tang LV, et al. Nanopore-targeted sequencing improves the diagnosis and treatment of patients with serious infections. mBio. 2023;14(1):e0305522. doi: 10.1128/mbio.03055-22.
- Yu G, Wang X, Zhu P, et al. Comparison of the efficacy of metagenomic next-generation sequencing and xpert MTB/RIF in the diagnosis of tuberculous meningitis. J Microbiol Methods. 2021;180:106124. doi: 10.1016/j.mimet.2020.106124.
- Wang J, Han Y, Feng J. Metagenomic next-generation sequencing for mixed pulmonary infection diagnosis. BMC Pulm Med. 2019;19(1):252. doi: 10.1186/s12890-019-1022-4.