
Abstract
Conversion of energy at the gas–solid interface lies at the heart of many industrial applications such as heterogeneous catalysis. Dissipation of parts of this energy into the substrate bulk drives the thermalization of surface species, but also constitutes a potentially unwanted loss channel. At present, little is known about the underlying microscopic dissipation mechanisms and their (relative) efficiency. At metal surfaces, prominent such mechanisms are the generation of substrate phonons and the electronically non-adiabatic excitation of electron–hole pairs. In recent years, dedicated surface science experiments at defined single-crystal surfaces and predictive-quality first-principles simulations have increasingly been used to analyze these dissipation mechanisms in prototypical surface dynamical processes such as gas-phase scattering and adsorption, diffusion, vibration, and surface reactions. In this topical review we provide an overview of modeling approaches to incorporate dissipation into corresponding dynamical simulations starting from coarse-grained effective theories to increasingly sophisticated methods. We illustrate these at the level of individual elementary processes through applications found in the literature, while specifically highlighting the persisting difficulty of gauging their performance based on experimentally accessible observables.
Graphical Abstract
1. Introduction: concepts & open questions
In chemical reactions at solid surfaces, different forms of energy are converted into one another. Chemical energy is released or consumed by breaking and making individual chemical bonds of adsorbed species or intermediates formed in the course of the reaction. Along the way, parts of this chemical energy are converted (transiently) into translational or vibrational energy of these surface species. Energy exchange can also occur with substrate degrees of freedom (DOFs), which (ultimately) will lead to a thermalization of adsorbates prevailing for a sufficiently long time at the surface. Net energy flow out of the adsorbate/surface fringe is thereby denoted as dissipation. Apart from (vibrationally excited) desorbing species, energy dissipation proceeds prominently into the substrate bulk. Largely, this occurs through the excitation of lattice vibrations, so-called substrate phonons [Citation1,Citation2]. At metal surfaces, however, the non-adiabatic excitation of electron–hole (eh) pairs provides another competing energy dissipation channel. The continuous distribution of electronic states across the Fermi level allows in principle to even excite lowest-energy eh-pairs – an argument that has often been used to suspect a high relevance of this channel at metal surfaces [Citation3–Citation7].
However, the real role and relative importance of both dissipation channels and how this varies across systems are presently largely unclear [Citation3,Citation8]. In fact, on this microscopic level we still know very little about these mechanisms of energy dissipation in general. This is rather intriguing, considering that chemical reactions at metal surfaces drive important applications and technologies like heterogeneous catalysis or surface growth. In the established microkinetic theories used in these fields [Citation9–Citation11] one, for instance, generally assumes that any reaction heat released in an exothermic reaction step is quasi-instantaneously dissipated away to ensure an immediate thermalization of the surface species. This motivates a Markovian view of an overall surface chemical reaction as a sequence of independent elementary steps such as adsorption, diffusion, reaction, and desorption as depicted in . Even though unanimously applied today [Citation12], the general validity of this Markovian picture is less clear from the microscopic perspective of the phononic and eh-pair dissipation mechanisms. In a typical exothermic surface reaction like dissociative oxygen adsorption at a transition metal surface heats of the order of a few electron volts (eV) are released. Considering that, e.g. the energy scale of phonons is meV, one may start to wonder how instantaneous the phononic energy uptake and concomitant thermalization of surface species really is.
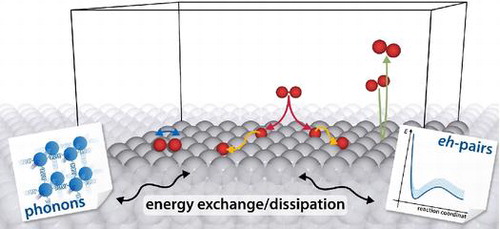
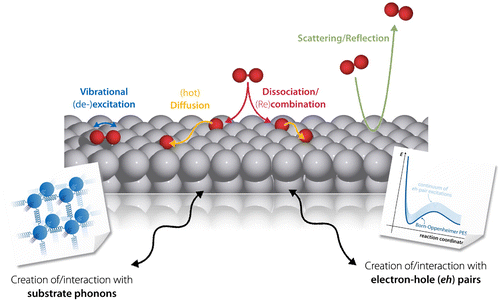
Figure 1. Various elementary processes in the context of GSD. An impinging molecule may, for instance, be directly reflected from the metal substrate, nevertheless exchanging energy and momentum with the surface. It may, however, as well dissociate on the surface – potentially through a vibrational precursor state – with the excess kinetic energy leading to hyperthermal motion of the fragments, so-called ‘hot diffusion’. In all of these processes, energy exchange with the metal occurring either through the excitation of lattice vibrations (phonons) or non-adiabatic electron–hole (eh)-pairs may significantly influence the resulting dynamics.
Additionally fueled by sustainability considerations concerning waste heat recovery or general heat management in heterogeneous catalysis, this has motivated fundamental research to arrive at a better microscopic understanding of energy dissipation at metal surfaces. Experimentally, the key approach are dedicated surface science experiments that study individual elementary processes at well-defined single-crystal surfaces in ultra-high vacuum [Citation13,Citation14]. The question of energy dissipation is thereby a sub-topic in the broader context of long-researched gas-surface dynamics (GSD). A central problem here is that key observables accessible in GSD experiments are often ensemble averages (vide infra). At present it is largely unclear in how much these observables are actually sensitive to details of the microscopic energy dissipation mechanisms. In fact, an unambiguous interpretation of the measured data is typically impossible without detailed modeling efforts. The dramatic increase in computer and algorithmic power has in this respect led to a strong surge of corresponding, in particular first-principles based theoretical work in recent years. This work has its own limitations though. It centrally still struggles with the necessity to simultaneously provide a reliable account of the surface electronic structure (and ensuing energetics) and adequately follow or sample the on-going surface dynamics. At the electronic structure level, the difficult task is to procure a description of localized orbitals of surface species on the one hand and the highly delocalized metallic band structure on the other [Citation15,Citation16]. Considering dynamical simulations, issues arise in turn from the extensive ensemble averaging required to compute key experimental observables, and from excessive system sizes when aiming to explicitly resolve the energy transfer to the multitude of electronic and phononic DOFs of an extended metal (Me) substrate.
These challenges can presently only be met by numerically efficient, effective theories. They typically rely on density-functional theory (DFT) with semi-local exchange–correlation functionals [Citation16–Citation20] and still often have to resort to bath-type treatments of the substrate DOFs. Open questions these theories try to address at the level of an individual elementary process (cf. ) include the relevance of each dissipation channel under specific conditions, whether these channels interact and influence each other or simply provide additive contributions, and how this picture will change from one system to another. Since all theories potentially able to consistently address these questions in practice are by nature approximate, validation by comparison to experiment is obviously vital. As such, another important aspect is also how experimentally accessible quantities in different types of GSD experiments at single crystal surfaces can be calculated. This not only from the perspective of reproducing these quantities accurately, but also to understand to which extent the details of the underlying energy dissipation mechanisms actually matter for the target properties of interest. In this topical review we survey the state-of-the-art of corresponding theories and their application in practice. For this we focus in subsequent sections on work done to elucidate the role of energy dissipation in adsorption processes (Section 2), in vibrational motion (Section 3) and in surface diffusion (Section 4). Emphasis is placed on concepts and in how much the impressive amount of work carried out in particular over the last years allows already to derive some general insights and ruling principles. An extensive list of references guides the interested reader to the methodological and technical details. The scope of this work is instead to introduce this lively field, its accomplishments and challenges to a broader audience.
2. Inelastic scattering & adsorption
Energy dissipation in adsorption processes is suitably studied in molecular beam experiments that expose an initially clean single-crystal surface to a beam of gas-phase species of defined initial kinetic energy and impinging from a defined initial angle. Detailed information about energy dissipation can then, e.g. be derived from an analysis of the translational and quantum state resolved vibrational and rotational energy distributions of inelastically scattered molecules [Citation14,Citation21]. For the ensemble of gas-phase molecules in a molecular beam, another central experimentally accessible kinetic quantity is the (initial) sticking coefficient, i.e. the fraction of molecules that has lost sufficient energy to remain adsorbed at the surface [Citation22]. The latter is usually a function of several variables such as beam incidence energy and angle, substrate temperature and specifically prepared rotational and vibrational quantum state of the impinging molecules [Citation23]. Sticking coefficients are thus beam-ensemble averages over the binary measure of whether an individual molecule adsorbs on a surface or not. So-gained information thus naturally convolutes effects of energy dissipation from several successive elementary processes that occur, e.g. during a dissociative adsorption process, such as the actual dissociation event, vibrational motion in a potential trapped precursor state, and subsequent diffusion of the reaction products (cf. ). As such, sticking coefficients are a prime example of GSD observables for which it is clear from the start that they will at best provide only very indirect insight into the detailed energy dissipation mechanisms. A substantial amount of first-principles modeling has nevertheless been devoted to compute these quantities. On the one hand, this is because high-accuracy references from experiment are available for a wide range of systems. On the other hand, sticking coefficients are fundamental kinetic quantities describing adsorption processes in microkinetic models. As such, it is most intriguing and relevant to understand which aspects of microscopic energy dissipation propagate through to this more coarse-grained, technological level.
2.1. GSD within the frozen surface approximation
Addressing sticking coefficients from a modeling perspective poses a significant challenge to theory. While impinging on the surface, the adsorbing species sample a wide range of configurations far away from the (ultimate) equilibrated adsorption geometry. This requires to accurately describe the potential energy surface (PES) representing the adsorbate–surface interaction over corresponding wide regions. Even for simplest diatomic adsorbates just accounting for the molecular DOFs (the position of the molecule’s center of the mass above the surface, its bond length and angular orientation) leads already to a six-dimensional PES that needs to be computed and represented. Explicitly treating at least all these molecular DOFs has thereby been established as a complete necessity by numerous studies which have specifically outlined the dangers of a reduced dimensionality treatment and shown how (intuition-based) simplifications over the adsorbate coordinates can yield dramatically wrong results with respect to extracted dynamical properties [Citation2,Citation24–Citation31]. A proper description of the beam-ensemble requires on top of this extensive statistical sampling of typically several tens of thousands of impinging molecular trajectories starting from varying initial conditions. A key concern thus foremost lies in procuring a numerically efficient, yet sufficiently accurate description of the aforementioned adsorbate–surface interaction potential.
The twofold nature of this challenge has promoted a likewise two-step, ‘divide-and-conquer’-like strategy [Citation25,Citation32–Citation34]: First, a continuous PES function is constructed by interpolating or fitting to a large set of ab initio data points that sample the adsorbate DOFs over a rigid, or so-called ‘frozen’ surface (FS) (depicted in blue in ). The evaluation of energies and forces on this continuous PES function then comes at a practically negligible computational cost which allows for extensive molecular dynamics (MD) simulations in a second step. While computationally tractable, at this FS level the inherent neglect of substrate mobility obviously precludes any (phononic) energy exchange with the lattice. Focusing for the time being also exclusively on the adiabatic Born–Oppenheimer PES, there is thus no energy dissipation mechanism explicitly considered in the model. Sticking must correspondingly be concluded by some ad hoc criterion, typically assuming a molecule to stick after its trajectory has prevailed for a sufficiently long time close to the surface or upon exceeding a threshold interatomic separation for the case of dissociative chemisorption.
Generally one would expect this crude approximation to work the better, the weaker the adsorbate couples to the lattice vibrations. Due to the large mass mismatch, this is best fulfilled for lightest H molecular adsorbates at transition metal surfaces, which for many reasons has been a prototypical GSD system [Citation35] that has received significant attention over the years anyway [Citation24–Citation28,Citation35–Citation37]. It is thus not altogether surprising that already FS simulations based on DFT-derived, six-dimensional PESs have been found to largely reproduce experimental H
sticking curves [Citation8,Citation37–Citation39]. An escalating activity is nevertheless being directed toward heavier and more strongly chemisorbing adsorbates in recent years. For these systems, in particular energy exchange with the lattice is expected to play an increasingly important role and requires that some (at least approximate) account of substrate mobility is included in the theory [Citation40,Citation41].
2.2. Effective models for phononic energy dissipation
Opening the phononic dissipation channel generally implies an exploding dimensionality for the problem at hand. Explicitly considering the motion of substrate atoms was hitherto largely prohibitive within the divide-and-conquer ansatz due to the increasing difficulty that is associated with each added DOF when constructing a continuous PES representation [Citation34]. This situation has just started to change as advanced, high-dimensional interpolation schemes such as the Behler–Parinello atomistic neural network approach [Citation42–Citation44] become increasingly applicable in the context of GSD [Citation45,Citation46]. As already conceivable from the simple Baule limit [Citation2,Citation14], however, energy transferred to the lattice in each direct adsorbate–surface collision is still small for light diatomic adsorbates. On this level, one can then tentatively consider the interaction with surface phonons to be more of a small perturbation to the FS-dynamics such that an explicit full-dimensional description of the lattice motion may not be altogether necessary.
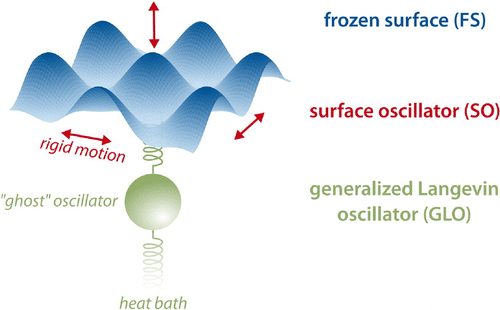
This motivates energy sink models that incorporate an effective treatment of surface mobility [Citation47–Citation53]. Such approaches generally target to reduce the phononic fine structure of the substrate into computationally convenient augmentations of the static-surface interaction in order to include some (approximate) account of energy exchange with the lattice. Motivated by an Einstein-like picture for the phononic system, the surface oscillator (SO) model [Citation47,Citation48] mimics a rigidly moving substrate by a single harmonic oscillator of assigned frequency and mass parameters. As shown in , coupling to the adsorbate is then straightforwardly described through a space-rigid shift in the FS expression for the PES. Extending on the latter as further depicted in , the generalized Langevin oscillator (GLO) additionally incorporates the approximate effect of a bulk thermal bath by coupling the SO reaction zone to an additional so-called ‘ghost’ oscillator [Citation47,Citation49–Citation53]. Energy dissipation and thermal fluctuations are consequently accounted for by subjecting the latter to frictional and random forces, respectively, rigorously satisfying the fluctuation–dissipation theorem in the numerical solution of the resulting generalized Langevin equation. While thus incorporating only a very limited number of additional DOFs, these models still allow to lift the FS-approximation by introducing the concept of a surface temperature with concomitant thermal motion [Citation1,Citation48] – even if it is just described in a coarse-grained way.
Figure 2. Hierarchical approaches to enrich simulations on a FS PES with an account of lattice motion. The SO model first introduces a 3D harmonic oscillator corresponding to a rigid shift of the entire PES in all spatial directions, thus allowing for adsorbate–surface energy transfer following a simple collision model. Building on this, the GLO adds a further ghost oscillator linearly coupled to the SO, which in turn is further coupled to a heat bath within an effective generalized Langevin description. This allows to also include energy dissipation from the SO to the bulk.
One of the main practical advantages of the SO/GLO approaches is their convenient application to any pre-existing adsorbate–surface PES that has already been derived on the level of the FS approximation. This also includes, of course, the continuous PES representations obtained from DFT within the aforementioned divide-and-conquer framework. In a first such application to H scattering from Pd(1 1 1) [Citation54], Busnengo and coworkers successfully used the SO model to reveal a channel of dynamic surface trapping that could explain the otherwise puzzling temperature dependence which was observed experimentally in the rotational excitation of scattered molecules [Citation55]. A similar result was reached only shortly afterwards also by Wang and coworkers for
scattering off Cu(1 1 1), but on the basis of quantum–mechanical (QM) wave-packet calculations [Citation56]. Opening also the GLO bulk dissipation channel as an outlet for the accumulating heat could further predict the low-temperature stabilization of a molecularly chemisorbed H
species at Pd(1 1 0) [Citation57]. While thus revealing important mechanistic details of the adsorption process, the inclusion of SO/GLO dissipation nevertheless provided only marginal corrections to averaged sticking probabilities for the light H
adsorbate [Citation58] (consistent with the previously mentioned good agreement with experiment already at the FS level). A much more dramatic effect was instead demonstrated for the heavier
molecule, during its highly exothermic adsorption at Pd(1 0 0) [Citation59]. Here, even a qualitative change in the shape of the calculated sticking curve was reported in comparison to the corresponding FS results, yielding thus a considerably improved agreement to experimental data [Citation60].
Despite these and many more successes (cf. e.g. Refs. [Citation61–Citation65]), it is nevertheless always prudent to bear in mind the inherent limitations that come with the underlying SO/GLO approximations. Recognizing foremost situations where these are bound to fail are cases where energy uptake cannot be reliably described on the level of only a single phonon excitation. Based mainly on energetic considerations, the frequency of the single SO oscillation is often taken to correspond to the surface (low-frequency, acoustic) Rayleigh modes, as these are generally assumed to be predominantly excited upon first impact with the surface (cf. e.g. Refs. [Citation66–Citation69] and references therein). The validity extent of this assumption, however, remains to its larger part unknown and could represent a highly dangerous over-simplification [Citation70,Citation71], in particular when going beyond prototypical studies of lightest adsorbates [Citation72–Citation75]. Even within a dominant one-phonon picture though, the SO model can only go as far as to provide a mechanical coupling of adsorbate–surface momentum exchange, i.e. accounting thus for energy loss to the substrate through lattice recoil and a back transfer to the adsorbate with surface temperature. Thereby considered ‘stiff’ lattice shifts will thus inherently fail to capture concomitant changes in the FS-PES that are induced by thermal displacements of the metal atoms from their equilibrium positions. This additional effect of vibrational surface motion can play an important role in the adsorption dynamics, as demonstrated prominently for (direct) dissociation at several metal surfaces [Citation76]. Here (as also in other reported instances [Citation77,Citation78]), DFT calculations specifically showed significant lowering of the activation barrier as a metal atom is puckered out of the surface at the transition state [Citation79,Citation80]. Effectively accounting for both of the aforementioned effects through two (independent) DFT-derived adsorbate–surface coupling parameters [Citation81], the quantum dynamics simulations of Jackson and coworkers thus arrived at a semi-quantitative agreement to experimental sticking curves (also reproducing the observed mode specificity and bond selectivity) that could not otherwise be achieved on the SO level alone [Citation82]. In conclusion, we note that this so-called barrier height modulation arising from thermal fluctuations of the lattice is expected to depend strongly on both the specific system and surface temperature under investigation, so that its overall relevance remains altogether a priori unclear in the context of other gas-surface reactions, as will be further discussed in the following.
2.3. Explicitly resolving surface motion
Direct ab initio molecular dynamics (AIMD) simulations stand fundamentally free of the aforementioned limitations discussed in the context of effective energy sink models. In this method the forces needed to propagate the equations of motion are full-dimensional Hellmann–Feynman forces computed from DFT ‘on-the-fly’ at each MD time step, so that no fitting or interpolation of the PES is required. Metal atoms are thereby treated explicitly within a fully mobile substrate and no a priori assumptions have to be made with respect to the heat bath Hamiltonian. This further naturally circumvents the extensive PES pre-evaluation required within the divide-and-conquer ansatz, but in turn comes at a tremendous increase in computational cost for each individual trajectory. With the advent of supercomputers and continuous developments toward improved algorithms for electronic structure calculations, this challenge is being steadily overcome and it is now increasingly possible to compute a meaningful number of AIMD trajectories for many systems that could not even be addressed in static calculations a few years ago [Citation83].
The combined dynamical picture of both adsorbate–surface coupling effects discussed previously (i.e. mechanical coupling of momentum exchange and barrier height modulation due to thermal fluctuations), should in principle be completely accounted for within AIMD simulations. Indeed, for the aforementioned dissociation (where both effects are known to be important [Citation76]) Kroes and coworkers predicted satisfying, semi-quantitative agreement to experimental reaction probabilities [Citation84]. In the meantime, similar results have also been reached for
[Citation37,Citation85,Citation86],
[Citation87,Citation88],
[Citation89], and C
[Citation78] adsorbates, overall showing AIMD simulations to provide a reasonable account of surface temperature effects. With reaction probabilities as the target observable, the effective advantage of the detailed AIMD account of adsorbate–surface energy transfer—over corresponding, numerically more attractive SO/GLO models—ultimately depends on the importance of thermal fluctuations (assuming of course the validity of the single-phonon approximation). As such, a simple GLO model has for example been shown to perform on an equal level as AIMD regarding calculated sticking probabilities for the dissociative
adsorption on W(1 1 0) [Citation90], while being entirely inappropriate for other systems such as, e.g. the aforementioned
on various transition metals [Citation76]. Unfortunately, however, such performance is extremely difficult to predict a priori, especially in view of the fact that both lattice coupling effects will induce qualitatively similar changes to the overall experimental sticking function with surface temperature.
At this point, however, it must be emphasized that the microscopic description of phononic dissipation as provided by present-day AIMD simulations is still far from perfect – a fact which stems (foremost) from practical, rather than conceptual, considerations. As the involved computational cost is largely determined by the substrate size, state-of-the-art AIMD studies are limited in practice to supercell setups involving slab models consisting of typically only a few surface lattice constants and layers [Citation83]. By construction, such models fail to provide the desired (high) resolution when representing the underlying phononic fine structure. For example the description of surface modes – which are assumed to be crucial for the initial energy uptake at the interface [Citation74] – will unavoidably suffer from the limited slab thickness that must be adopted in computationally tractable DFT supercells. Most importantly, the imposed periodic boundaries (which are essential to describing the underlying metallic band structure) will restrict any phonon propagation within the finite extent of the latter, thereby leading to an unphysical confinement of the released chemical energy. Largely exothermic reactions in particular are thus increasingly prone to significant overheating of the metal substrate – an effect which severely compromises the description of the ensuing equilibration process and may even critically modify the actual adsorbate dynamics, as will be discussed in more detail in Section 4.2. While the effect of these shortcomings has not been investigated in detail, they may still be responsible for remaining experiment–theory discrepancies found in literature. A prototypical such example is the dissociation of HCl on Au(1 1 1) for which energy dissipation is believed to play a key role [Citation91]. While (independently performed) AIMD simulations [Citation86,Citation92] have consistently overestimated the experimental probabilities [Citation91], the corresponding reaction dynamics of this system remain an enigma and have become quite a controversy in the field [Citation86].
An explicit treatment of substrate mobility within a (suitably) sizable heat bath thus continues to pose a big challenge to contemporary AIMD simulations and thereby derived accounts of phononic dissipation. This situation is aggravated by the extensive statistical sampling that is hereby required not only for describing the beam-ensemble, but also the thermal distribution of metal atoms at finite surface temperatures. With current computational resources typically providing up to ‘only’ a few hundred trajectories, AIMD-derived statistics are thus presently still very poor in particular when addressing rare events. In this respect, using existing AIMD configuration sampling as the basis for parameterizing the aforementioned upcoming high-dimensional neural network-based interpolation schemes [Citation42–Citation44] represents a promising alternative for the future that will allow for routinely evaluating several orders of magnitude more trajectories as compared to further explicit AIMD simulations [Citation45,Citation46]. In the meantime, while entirely neglected so far, the channel of non-adiabatic energy dissipation adds yet another facet of complexity to GSD and toward which attention is directly turned in the following.
2.4. Exciting electron–hole pairs: non-adiabatic effects
Energy dissipation arising from non-adiabatic effects, i.e. the dynamical coupling of the adsorbate nuclear motion to excitations in the metallic continuum of electronic states at the Fermi level, still holds a rather suspect role in gas-surface scattering/adsorption processes [Citation3–Citation7]. One of the most controversial showcases in this respect is the highly exothermic oxygen dissociation at Al(1 1 1) that proceeds through an triplet-singlet spin transition as the molecule approaches the surface [Citation8,Citation74]. Here, extremely low initial sticking coefficients measured for small
incidence energies are strongly indicative of activated adsorption – a process which goes entirely amiss within the essentially barrierless (adiabatic) PES that is evaluated on the level of the DFT generalized gradient approximation (GGA) [Citation93]. It remains, however, altogether unclear whether this actually indicates a hindered triplet-singlet transition due to spin selection rules that would give rise to corresponding dynamical barriers. Alternatively, this omission could instead be rooted in shortcomings of the employed semi-local exchange–correlation functionals [Citation74,Citation94,Citation95]. Another prominent example vouching for pronounced non-adiabatic effects is the energy loss associated with vibrationally excited NO scattering from Au(1 1 1) [Citation7,Citation96–Citation98]. The non-adiabatic relevance is strongly suggested here by multi-quantum relaxation of the vibrating molecule that is clearly observable on the metal, but completely absent on an insulating LiF surface [Citation97].
Going beyond the Born–Oppenheimer approximation in order to account for such non-adiabatic effects requires in principle the full propagation of a combined, high-dimensional nuclear-electron wave function that would naturally also include the multitude of potential electronic excitations. Such an endeavor, however, is computationally out of reach in the context of GSD for at least decades still to come. Including electronically non-adiabatic effects in computational simulations thus inevitably requires further approximations and simplifications leading to an effective treatment, such as applying a mixed quantum–classical description of the system in terms of a mean field Ehrenfest [Citation99–Citation102] or surface hopping framework [Citation99,Citation103–Citation106]. The latter has, for instance, been successfully invoked to explain the aforementioned NO vibrational de-excitation on Au(1 1 1) through an electron-transfer mechanism [Citation105,Citation107,Citation108]. Further coarse-graining the electron dynamics can be achieved on the basis of time-dependent perturbation theory [Citation109–Citation111]. Counting among the disadvantages of all these methods is that they either remain computationally very intense, or require extensive parametrization, or impose specific symmetry constraints on the simulated trajectories such that a general applicability to routinely performed gas-surface calculations is lost.
Presently the only viable solution for large-scale gas-surface simulations thus lies in entirely replacing the electronic degrees of freedom within the concept of electronic friction [Citation112–Citation114]. As formally shown by Tully and Head-Gordon [Citation114] – and recently more rigorously also by Dou and Subotnik [Citation115–Citation117] – the idea behind this approach is to start with a mixed quantum–classical description of the dynamical (electron-nuclear) system. Following a suitable Miller-Meyer action-angle transformation, the explicit electronic DOFs are subjected to a generalized Langevin replacement that is valid within the weak-coupling approximation and results in a generalized Langevin equation for the nuclear DFOs. This, however, is not the form of electronic friction theory that is used in practical MD simulations today. Hence, in a second step the explicit time-dependence of the friction kernel (collectively condensing the dynamics of the electronic DFOs) is removed by invoking a Markov approximation in the electronic subspace implying (infinitely) short electronic coherence times [Citation111]. Thus assuming memoryless eh-pairs that immediately ‘forget’ about the past yields ultimately a Langevin equation for the nuclear dynamics that combines all non-adiabatic effects in a single electronic friction coefficient. Altogether, the electronic friction approach thus allows to augment the Born–Oppenheimer ground-state dynamics with a dissipative friction force and a temperature-dependent fluctuating force, as manifested through the fluctuation–dissipation theorem. This combination of conceptual simplicity and numerical efficiency, along with the possibility of application to dynamics on a potentially pre-calculated Born–Oppenheimer PES has overall served to the great popularity of this method.
While the idea of including frictional energy losses into nuclear dynamics dates back to more than 30 years ago [Citation112,Citation118–Citation122] it was not until much more recently that Juaristi and coworkers adopted this concept for routine DFT-based applications within the divide-and-conquer framework [Citation123]. The key ingredient hereupon relied on is an approximate construction of the required molecular friction tensor from independent contributions of individual atoms. As depicted in , these are estimated from a simple jellium-based embedding model [Citation118,Citation119,Citation124,Citation126] to finally yield isotropic atomic friction coefficients as a function of an embedding electronic density. Within this so-called local density friction approximation (LDFA) [Citation113,Citation123], the latter is taken as the local electron density of the clean metal surface at the position of the (individual) adsorbate atoms. This simplification consequently allows for a straightforward interpolation of the friction coefficients into a convenient analytical function of the electronic density only. This distinguishes the LDFA from more refined orbital-dependent friction (ODF) methodology [Citation114,Citation122,Citation127,Citation128] whose numerical involvement [Citation128–Citation130] (until only very recently [Citation131]) has hindered application to high-dimensional models though [Citation132–Citation137].
Figure 3. Construction of electronic friction coefficients within the LDFA [Citation123]. The interacting molecule-surface system is first approximated through independent atoms being embedded in the electron density of the clean metal surface. The local electronic density at the atomic positions is then used to independently map to an isotropic atomic embedding model system of a spherically symmetric impurity in jellium. Finally, electronic friction coefficients are evaluated from the scattering phase shifts of the Kohn–Sham states at the Fermi-momentum for this model [Citation118,Citation119,Citation124,Citation126]. This ultimately yields, for each element, an electronic friction coefficient as a function of the embedding density and can thus be conveniently evaluated and tabulated prior to dynamical simulations. It has later been suggested to introduce molecular information by constructing the embedding density via a suitable Hirshfeld-partitioning [Citation140] of the full system electronic density [Citation141].
![Figure 3. Construction of electronic friction coefficients within the LDFA [Citation123]. The interacting molecule-surface system is first approximated through independent atoms being embedded in the electron density of the clean metal surface. The local electronic density at the atomic positions is then used to independently map to an isotropic atomic embedding model system of a spherically symmetric impurity in jellium. Finally, electronic friction coefficients are evaluated from the scattering phase shifts of the Kohn–Sham states at the Fermi-momentum for this model [Citation118,Citation119,Citation124,Citation126]. This ultimately yields, for each element, an electronic friction coefficient as a function of the embedding density and can thus be conveniently evaluated and tabulated prior to dynamical simulations. It has later been suggested to introduce molecular information by constructing the embedding density via a suitable Hirshfeld-partitioning [Citation140] of the full system electronic density [Citation141].](/cms/asset/2e0ef15b-7cfc-4aef-a3cd-c19666748969/tapx_a_1381574_f0003_oc.gif)
The inherent assumption of independent atoms within the original LDFA approach, however, triggered a controversial discussion [Citation123,Citation138,Citation139], mostly because it ignores the adsorbate’s molecular character and thus lacks, for instance, the steep increase of friction coefficients at dissociative transition states predicted from ODF [Citation134,Citation135]. In this regard, it has recently been suggested to introduce molecular information through suitably Hirshfeld-partitioned [Citation140] full system densities to map to the LDFA model system [Citation141] rather than relying on clean metal densities (vide infra). Moreover, one may pragmatically argue that the immanently low velocities in such transition state regions effectively suppress the contribution of the velocity-scaled friction term within the actual dynamics and thus wash out potential inaccuracies [Citation123] – at least when focusing on reaction probabilities [Citation131].
Retaining the LDFA within the independent atom approximation and its unique numerical efficiency, many divide-and-conquer studies have been revisited over the past years to augment the existing FS-based MD simulations with electronic friction. Alducin and coworkers specifically investigated the dissociative adsorption of several small molecules such as [Citation123],
[Citation123,Citation142],
O [Citation143] and
[Citation144] on various metal surfaces. The overall conclusion drawn from these studies is that the non-adiabatic energy dissipation channel produces only small effects on calculated sticking probabilities. Alone a full dimensional account of the adsorbate PES was in fact found to alleviate certain discrepancies with respect to experiment that had previously been erroneously assigned to non-adiabatic effects through a low-dimensional ODF-description. Prominent such examples are
adsorption on Ru(0 0 0 1) [Citation134] and the vibrational de-excitation of
scattering from Cu(1 1 1) [Citation37,Citation38,Citation135,Citation145].
Similar to what has already been discussed in the context of phononic dissipation, however, the overall insensitivity reported for averaged sticking coefficients does not necessarily extend to mechanistic details of the underlying adsorption/scattering process. Conclusions along these lines have, e.g. already been independently reached for the dissociative adsorption on Ru(0 0 0 1) [Citation146] and Ag(1 1 1) [Citation131]. Here, the authors specifically noted minor non-adiabatic energy losses that may be unimportant to reaction probabilities, yet significantly influence other experimentally accessible observables such as the energy distribution of backscattered molecules. This is of particular interest considering the tensorial character of the electronic friction that is – in contrast to LDFA – directly accessible from ODF [Citation114,Citation134]. In this regard, recent studies by Maurer and coworkers [Citation127,Citation128,Citation131] actually demonstrated that hitherto often neglected contributions are indeed rather pronounced and may, besides the well-known mode-specific electronic friction [Citation127,Citation128,Citation147,Citation150], lead to dynamical steering and vibrational mode coupling effects. As a concluding remark, it must be emphasized though that the popular electronic friction approach may even be altogether inappropriate for generally gauging the relevance of this dissipative channel. This is due to its inability to describe strong non-adiabatic coupling, as has, e.g. been most prominently shown for the multi-quantum vibrational transitions occurring during NO scattering on Au(1 1 1) [Citation151]. Notwithstanding, at present electronic friction is the best shot we have at all to address non-adiabatic energy dissipation in high-dimensional surface simulations.
2.5. Combining phonons and electron–hole pair excitations
Combining the two dissipation channels in dynamical simulations promises illuminating insight into the relevant importance of each channel, at least on the specific level of the theory employed. Unfortunately, such applications have hitherto been rather scarce. In the late 1990s Tully and coworkers were the first to combine the electronic friction approach with an explicit description of lattice degrees of freedom for the scattering of CO from Cu(1 0 0) [Citation152]. In this early study, however, the authors relied on a semi-empirical potential for describing the adsorbate–metal interaction and no supporting experimental data were available at the time. A dominating role of the phononic dissipation channel was reported that outperformed the non-adiabatic energy losses by up to a factor of ten (depending on the CO incidence energies). Similar findings were also reported much more recently for the scattering of nitrogen atoms and molecules using a combined electronic friction/GLO model applied to DFT-derived continuous PES models [Citation61,Citation62,Citation153,Citation154]. Here, phononic effects were found superior over the non-adiabatic counterpart on essentially every measure that was considered. This said, most qualitative aspects of the dynamics could admittedly already be reached on the level of a purely adiabatic FS model when accounting for all adsorbate DOFs.
Quite in contrast, for somewhat lighter (atomic) adsorbates, an outstanding importance of eh-pair excitations has been reported. Evaluating frictional non-adiabatic energy losses non-self consistently from AIMD trajectories, Kroes and coworkers predicted that the latter exceeds phononic energy losses by factor of 2.5 and 6 for H atom scattering off Cu(1 1 1) and Au(1 1 1), respectively [Citation155]. This picture was later substantiated by Wodtke and coworkers who reported a pronounced disagreement between experimentally measured energy-loss spectra for backscattered H atoms from Au(1 1 1) and adiabatic simulations on a carefully parametrized full-dimensional PES based on effective medium theory including all surface DOFs [Citation156–Citation158]. Adding LDFA-electronic friction forces, however, resulted in a spot-on agreement with experimental measurements. Finally, a larger relevance of the non-adiabatic against the phononic dissipation channel was also reported for the recombination of as compared to
via the Eley–Rideal abstraction mechanism (i.e. involving direct collision and recombination with an adsorbed species from the gas phase) [Citation159,Citation160]. Altogether it nevertheless remains doubtful whether these findings actually reflect an absolutely increased relevance of non-adiabatic energy losses. The increased relative relevance compared to phononic channel could also merely arise from a suppression of efficient phononic dissipation for these lightest adsorbates.
3. Trapped at the surface: gauging the tools for vibrational damping
Vibrations of molecular adsorbates that are ‘trapped’ at the metal surface can, in principle, decay via coupling to substrate phonons, and/or through the electronically non-adiabatic excitation of eh-pairs [Citation3]. In addition, vibrational energy may flow to other internal molecular modes, adsorbate–surface vibrations, and even (sufficiently close) neighboring species. In disentangling the contributing role of numerous such mechanisms, it is useful to consider corresponding coupling strengths in terms of smaller or larger mismatch in timescale. High-frequency adsorbate vibrations are, for example, expected to be predominantly relaxed through eh-pair excitations, simply due to their large frequency mismatch with all other vibrations present [Citation150,Citation161]. Advocated by Persson and Persson already more than thirty years ago [Citation122,Citation162–Citation167], this notion was further substantiated by Tully and coworkers by investigating the C–O stretching mode of CO on Cu(1 0 0) [Citation168]. In their study, agreement to the experimentally measured vibrational lifetime of about 2 ps [Citation169] could only be achieved by including dissipative non-adiabatic effects in terms of electronic friction, which lowered the corresponding adiabatic prediction by a whopping six orders of magnitude. More recently, Saalfrank and coworkers reported similar findings for the -mode (frustrated translation perpendicular to the surface) of adsorbed hydrogen atoms on Pb using explicit high-level AIMD plus electronic friction (AIMD+EF) simulations [Citation170].
Vibrational damping of high-frequency adsorbate modes thus provides the idealized ‘isolated’ setting for gauging the accuracy of non-adiabatic theories in GSD. The process is (at least, largely) dominated by a single dissipation channel and atoms are confined to the vicinity of their energy minima, thereby eliminating the need for exploring vast PES regions as required for modeling gas-phase impingement. Most importantly, vibrational lifetimes provide a direct measure for the rate of energy flow and are accurately accessible as benchmark observables from real-time experimental measurements using e.g. pump-probe spectroscopy [Citation150].
Based on this foundation, a plethora of computational studies focused on reproducing experimentally measured vibrational lifetimes for small molecular adsorbates on metal surfaces using different underlying models for the electronic friction coefficients [Citation127,Citation128,Citation141,Citation147,Citation149,Citation171]. In detail, Persson and coworkers [Citation149] as well as Tully and coworkers [Citation127,Citation128,Citation147,Citation148,Citation171] evaluated elements of the ODF friction tensor along certain normal modes for CO [Citation127,Citation128,Citation147,Citation149,Citation171], CN [Citation128,Citation148,Citation149] and NO [Citation128,Citation148] on various coinage and transition metals. Given the numerical challenges involved in adequately sampling the underlying metallic band structure and evaluating the required matrix elements to arrive at ODF friction coefficients [Citation128,Citation129,Citation148], the reported values emerging from various implementations and numerical setups show considerable spread. Notwithstanding, in particular for highest-frequency (and thus anticipated to be ‘most non-adiabatic’) adsorbate stretch modes, calculated vibrational lifetimes were generally found to agree with experiment to within the same order of magnitude. The computed non-adiabatic decay rates further illustrated a pronounced mode-specificity that, at least so far, eludes any clear, system-transferable trend [Citation127,Citation128,Citation148,Citation171]. Unfortunately, an unambiguous experimental verification of this prediction remains unattainable, in particular for low-frequency modes, as the latter are more likely to couple to surface phonons and thus again open Pandora’s box of competing dissipation mechanisms.
Confined therefore to investigating only high-frequency adsorbate stretch modes, a more recent benchmark study [Citation141] focused on assessing the performance of the popular LDFA approach [Citation113,Citation119,Citation123,Citation125]. As mentioned before, electronic structure information here only enters on the level of an atomic embedding density. Given this simplicity, it is rather surprising to note that dynamically evaluated vibrational lifetimes obtained with LDFA-based friction coefficients were found to perform on an equal level as previous ODF-based studies for several experimentally well-characterized systems [Citation141]. A recent study suggested this good performance of the LDFA to arise from finite electronic coherence times, which wash out details of the electronic band structure that are in any case neglected in the LDFA [Citation111]. Further work is, however, required to better assess the full validity and performance of the LDFA. As a pragmatic bottom line, at least for the time being it seems that both the very simple LDFA approach as well as the computationally more demanding ODF model account reasonably well for the non-adiabatic energy losses of high-frequency stretch modes of molecular adsorbates on metal surfaces. Yet, as appealing as it may be to interpret this performance as a justification to rely on these models also for other surface dynamical processes, a respective generalization has to be taken with considerable caution. Benchmarking against vibrational lifetimes avoids delicate situations encountered in simulating, e.g. adsorption processes such as a steep increase of friction coefficients at dissociative transition states [Citation123,Citation131,Citation134,Citation135,Citation138,Citation139] or singularities in the latter at spin transitions [Citation132,Citation133,Citation172]. However, these are situations, that anyhow question the limits and validity of the electronic friction approach in general, not only the LDFA [Citation114–Citation117,Citation172].
4. Surface diffusion
Similar to the vibrational motion of adsorbed species, on-surface diffusion is largely controlled by the rate of energy exchange with the underlying substrate [Citation173,Citation174]. However, here no striking argument (such as the frequency mismatch detailed in the previous section) can be invoked in order to safely disregard either one of the available microscopic dissipation channels. Quite on the contrary. Good reasons can be brought forward to argue in favor of both channels: Long adsorbate–surface contact times and the concomitant high embedding densities are suggestive of non-adiabatic coupling within an electronic friction-based description, while the relatively long time scales that are characteristic of diffusive motion should intuitively favor a more efficient phononic dissipation channel.
4.1. Thermal diffusion
For a long time the classical notion was that once thermalized with the surface, energy exchange during diffusion of surface species happens predominantly through phononic coupling [Citation173]. This picture was also nurtured from a pioneering study by Tully and coworkers who showed that electronic friction forces do not significantly influence the equilibrium diffusive motion of CO on Cu(1 0 0) [Citation152]. Notwithstanding, even the authors themselves remarked at the time that the absence of a notable influence on the diffusion rate in this study neither necessarily implies a non-existent coupling to eh-pairs nor any generality of their findings. Instead, their description of the phononic bath through harmonic oscillators may have just provided an energy sink effective enough to inhibit correlated multi-jump diffusive motion, such that additional non-adiabatic damping effects could be masked when focusing only on diffusion rates. This is in line with findings by Wahnström. These suggested a dominant role of eh-pair excitations over very inefficient phononic couplings for the diffusion of H on Ni(1 0 0) that is indeed found to proceed via correlated jumps [Citation175].
Similar to the situation for adsorption processes, more sensitive observables than mere diffusion rates thus seem to be necessary to develop further insight into the role of the dissipative channels. Along this line of thinking, Rittmeyer and coworkers recently analyzed Helium-3 spin echo (He-SE) measurements [Citation176], which provide time-resolved access to the surface (auto-)correlation function. With the corresponding decay rates very sensitive to the adsorbate–substrate coupling and the underlying diffusion mechanism [Citation177–Citation179], the authors evaluated the non-adiabatic contribution to these rates within the LDFA approach. Intriguingly, a high degree of non-adiabaticity suggested a more pronounced role of eh-pair excitations than anticipated by the classical ‘textbook notion’, at least for this system.
4.2. ‘Hot’ diffusion
The prevalent notion of thermal diffusion presupposes the adsorbate’s continuous equilibration with the surface – regardless of the detailed origin of the coupling – and correspondingly predicts substrate temperature as the ruling factor. This is contrasted by the concept of hyperthermal transient mobility which can arise from the intrinsic exothermicity of an immediately preceding elementary step like (dissociative) adsorption. Non-instantaneous thermalization of the released chemical energy drives the ensuing ‘hot’ adsorbate diffusion that is then governed by dissipation to the underlying surface, rather than the substrate’s overall temperature. This transient mobility thus intricately couples the elementary reaction steps of adsorption and diffusion – an implication hitherto not considered in the present-day Markovian microkinetic formulations in surface catalysis [Citation9,Citation10]. Concepts embracing such processes are nevertheless becoming increasingly established over the past decades [Citation180]. Ensuing for example the (exothermic) dissociative oxygen adsorption, so-called ‘hot’ adatom motion has been persistently reported by scanning tunneling microscopy (STM) experiments for several metal surfaces [Citation181–Citation187]. Working at temperatures that are sufficiently low to suppress thermal diffusion, corresponding STM studies infer hyperthermal diffusion from recorded larger or smaller separation distances between adatom pairs after equilibration – an indirect procedure that has in fact caused quite some controversy [Citation188–Citation190].
From a modeling perspective, the accuracy of energy sink models in describing such hypterthermal motion is challenged head on as the entire process hinges on the rate of dissipation to the substrate. Such a sensitive dependence was already demonstrated in the aforementioned seminal work of Tully and coworkers for hyperthermal CO migration on Cu(1 0 0) [Citation152]. Including electronic friction in classical MD simulations was found to significantly quench the molecules’ on-surface transient mobility – an effect which did not extend to the case of equilibrium diffusion. A similar conclusion was recently reached for hot diffusion of atomic H on a precovered W(1 1 0) surface [Citation191]. As a result, the Eley–Rideal reaction was predicted as the more likely mechanism for abstraction over hot-atom recombination/desorption. On the other hand, Wahnström and coworkers estimated the eh-pair dissipation mechanism to be relatively unimportant for the hyperthermal adatom motion following the dissociative
adsorption at Al(1 1 1) [Citation175,Citation192]. Even when focusing solely on phononic damping within a Langevin framework, however, corresponding simulations showed no evidence of a high transient mobility that could reconcile the exceptionally large O–O separation distances derived from STM experiments [Citation181]. Lateral displacements were instead found to be primarily limited by the high PES corrugation and concomitant rapid randomization of the adatom motion, while adsorbate-phonon coupling was overall reported to be weak. At this point, however, it is worth noting that the semi-empirical potentials used in this work to describe the O-Al interaction could not be extended to modeling the actual (preceding)
dissociation event, thereby prohibiting a realistic sampling of the employed initial conditions. Unless there is a problem in the STM interpretation [Citation188,Citation189], this obvious limitation represents an equally likely cause for the discrepancy to experiment as the approximate treatment of adsorbate–surface energy transfer.
Overcoming this limitation, a pioneering study of Groß employed AIMD to consistently model dissociation and the subsequent adatom thermalization at Pd(1 0 0) [Citation19]. Based on substrates of about 100-200 metal atoms, this predicted ‘hot’ H adatoms that transiently diffuse to an average separation distance of three to four surface lattice constants. Adding an LDFA-based electronic friction to this description, Blanco-Rey and coworkers later on arrived at halved H–H separations only [Citation193]. The authors consequently advocated a dominating role for the electronic dissipation channel arguing an efficiency of about five times larger than provided by the phonon bath [Citation193]. Subsequent work in this direction [Citation194,Citation195] specifically highlighted the increasing contribution of eh-pair excitations during the later stages of the relaxation process (i.e. for long adsorbate–surface contact times), even if non-adiabatic effects can be neglected during the preceding dissociation event [Citation110,Citation123,Citation134,Citation146,Citation196]. At this point, however, it has to be noted that in present-day combined AIMD+EF simulations, the effective
-Langevin description of the non-adiabatic bath provides an infinite energy sink, thus conflicting with an explicitly described, energy conserving lattice motion.
As already mentioned in Section 2, even self-standing AIMD (and AIMD+EF) simulations remain challenged in providing an altogether satisfactory reference for phononic dissipation. Computationally tractable slab models compromise the description of the phonon band structure and limit their propagation to a finite volume of few substrate atoms in each direction. This gains particular relevance in the present context of hyperthermal diffusion by considering that a (preceding) exothermic surface reaction may easily release several electronvolts of energy. Unphysical phonon reflections at the periodic boundaries of the employed supercell may thereby quickly lead to severe substrate ‘overheating’, while falsifying the ps-scale equilibration of the actual adsorbate dynamics [Citation197].
In solving this problem, the recent development of the QM/MM embedding approach for metals (originally coined, and hence referred to, as ‘QM/Me’) [Citation197] represents a big step forward. Here, a DFT-based treatment of the immediate reaction zone (i.e. typically around the adsorbate impingement site) is complemented by an extended substrate that is described on the level of a many-body classical interatomic potential. The latter are generally sufficiently accurate in providing a realistic, material-specific representation of lattice deformations (cf. e.g. Refs. [Citation198,Citation199]), while their numerical efficiency allows for capturing all associated long-range elastic effects. Within a multi-scale modeling philosophy, QM/Me thus augments standard AIMD with a fully quantitative account of phononic dissipation as heat flows from the embedding region and into the macroscopic metal bath, as schematically illustrated in .
Figure 4. Schematic illustration of the QM/Me embedding approach, applied here to the dissociative adsorption over Pd(100) [Citation190]. A QM description of the immediate reaction zone is based on periodic DFT calculations to yield an accurate description of the short range, adsorbate-induced chemical interactions. This QM-cell is embedded into an Me substrate that is treated at the level of a numerically efficient classical interatomic potential to provide the elastic contribution to the forces due to deformation of the lattice. Released chemical energy is thus dissipated out of the ‘hot’ reaction zone and into a ‘cold’ macroscopic heat bath, while atoms in the embedding cell are color-coded according to kinetic energy.
![Figure 4. Schematic illustration of the QM/Me embedding approach, applied here to the dissociative adsorption over Pd(100) [Citation190]. A QM description of the immediate reaction zone is based on periodic DFT calculations to yield an accurate description of the short range, adsorbate-induced chemical interactions. This QM-cell is embedded into an Me substrate that is treated at the level of a numerically efficient classical interatomic potential to provide the elastic contribution to the forces due to deformation of the lattice. Released chemical energy is thus dissipated out of the ‘hot’ reaction zone and into a ‘cold’ macroscopic heat bath, while atoms in the embedding cell are color-coded according to kinetic energy.](/cms/asset/1b36d059-e8e5-4d64-a09f-7143f368845b/tapx_a_1381574_f0004_oc.gif)
In a first application to oxygen dissociation over Pd(1 0 0), QM/Me showed phonons dissipating the vast majority of the released chemical energy into the bulk, i.e. outside of the QM-cell, already within ca. 1.5 ps after the initial bond breaking [Citation197]. The observed ps-scale rate of heat transfer to the substrate nevertheless clearly demonstrated that this process is not instantaneous on the timescale of the actual adsorbate dynamics. Furthermore, the predicted equilibrium O–O separation distance of four surface lattice constants could not be reproduced within ‘pure’ AIMD simulations, thus underscoring the importance of an extended and explicit description of the surface degrees of freedom for microscopic details. Analyzing the role of surface symmetry, a subsequent QM/Me study compared this result for different Pd facets to reveal a striking difference in the transient lifetime of the hyperthermal O state [Citation75,Citation190]. More specifically, a much slower equilibration was found on Pd(1 1 1) that would seemingly contradict the shorter O–O end distances previously reported from STM measurements [Citation185]. Relying on the atomic resolution of the QM/Me heat bath allowed for rationalizing this finding through a mode-specific analysis of corresponding phonon excitations: This identified the dominant dissipation channels as qualitatively different groups of localized surface modes that do not necessarily involve a predominant Rayleigh excitation, as principally assumed for energy sinks in model bath Hamiltonians (cf. e.g. Refs. [Citation68,Citation69,Citation200,Citation201] and references therein). Instead, the complex adsorbate-phonon dynamics give rise to a sensitive dependence on details of the phononic fine structure that may lie on either the high- and/or low-frequency end of the spectrum. This can ultimately lead to intrinsically different rates of dissipation to the bulk that would otherwise go entirely unnoticed from the perspective of experimentally accessible product end distances.
5. Summary and conclusions
In the present review we set out to provide a comprehensive overview on modeling efforts for describing the conversion of energy at the gas–solid interface. Focusing on technologically relevant metal surfaces, we consistently address energy dissipation through the (competing) excitation of substrate phonons and eh-pairs at the level of individual elementary processes. Principal questions of focus concern the appropriate tools for modeling each of these microscopic dissipation channels, their effective role in driving the dynamics, and how this is ultimately reflected in (experimental) target observables of interest. Throughout the review, we report on numerous studies aimed at answering these questions, but note that we do not even come close to a full reference list for this highly active field. While thus demonstrating the considerable progress that has been made over the past few decades, we specifically highlight the persisting difficulty of gauging the performance of the various theoretical tools that have been proposed or, vice versa, the level of detail that is required to be accounted for. While each of the discussed methods comes with its own limitations and challenges, this problem stems mainly from the largely inconclusive picture drawn on the basis of different experimental target quantities and across different systems.
This is, for example, clearly demonstrated for the processes of inelastic scattering/adsorption reviewed in Section 2. Here, the importance and ubiquity of reaction probabilities (i.e. sticking coefficients) as measured from molecular beam experiments have typically provided a key measure to compare against. Attempts to reproduce such sticking data from theory has shown that dissipation from phonons can require in some cases an explicit treatment of substrate motion, while satisfactory agreement is often already achieved on the level of effective energy sink models, or even a complete neglect of energy exchange with the lattice. While some prediction of the phononic relevance can be made based on the adsorbate–substrate mass mismatch, this does not always appear to be accurate [Citation78,Citation86,Citation92]. This uncertainty adds yet another facet of complexity when considering also the contributing role of the non-adiabatic dissipation channel, whose relevance remains to date similarly obscure and inconclusive. It must be noted, however, that our judgment here is likely further clouded by the limited accuracy of approximate exchange–correlation functionals within DFT. With the vast majority of studies in GSD still relying on GGA-type functionals for computational convenience and clearly no universally ‘best’ functional at sight, these inaccuracies are particularly manifested in the calculation of encountered energy barriers [Citation37,Citation38]. As reaction probabilities very sensitively depend on the latter, it could even be that effective dissipation models are simply ‘compensating’ for incorrect barrier heights. Experimental sticking coefficients thus overall represent a rather inappropriate measure for gauging the accuracy of dissipation models: They convolute many different effects along the trajectory course of impinging gas-phase species, while their stochastic nature prevents a sensitive response to microscopic details of the dynamics which may even ‘wash out’ all signatures of phononic and/or non-adiabatic dissipation. Alternative experimental measures such as the, potentially state-resolved, energy distribution of backscattered molecules thus appear as more promising benchmark references [Citation131].
Also in this respect, the vibrational damping studies addressed in Section 3 are of great value. Here, only a limited phase-space region of the PES is encountered during the dynamics, the non-adiabatic dissipation channel provides a clearly dominating contribution and, most importantly, detailed, dissipation-specific experimental support is made available from pump-probe measurements. Thereby derived vibrational lifetimes have thus provided an extremely sensitive measure to non-adiabatic effects and have largely served as benchmark observables in establishing the performance of the numerically efficient electronic friction approach. Notwithstanding, the extent to which this trust can be transferred also to other elementary processes remains mostly unclear, especially when accommodating the adsorbate’s description at a (reaction) transition state during the making/breaking of chemical bonds. One crucial requirement in this regard is the validity of the underlying physical picture that implies only weak non-adiabatic coupling. Despite its limitations, however, electronic friction presently remains the only tractable method in terms of computational resources needed for extended systems and times scales also relevant to phononic motion. As such, already this assessment as extracted from studies of vibrational damping, even if not necessarily universal, is of great importance.
Generally establishing such trust is very important also for simulating surface diffusion as discussed in Section 4. Here, long adsorbate–surface contact times call for all the more accuracy in describing energy exchange with an atom-resolved substrate. Microscopic details are, in principle, hereby expected to gain increasing importance for phonon effects (showing, e.g. a pronounced dependence on surface symmetry [Citation75,Citation190]) and eh-pair exciatations [Citation195] alike. However, the relevance of the two dissipation channels is again here far from obvious. Unfortunately, only very few first-principles diffusion studies have so far simultaneously included both of these contributions, while existing indications support a more pronounced role for non-adiabatic couplings than hitherto anticipated [Citation173]. In this respect, further comparisons against high-quality, atomically resolved experimental data such as, e.g. provided from STM-derived adsorbate end-distances or He-SE signatures will undoubtedly play a key role in systematic future studies. Largely removing uncertainties with respect to the phononic heat bath, the QM/Me embedding scheme (described in Section 4.2) now represents a promising way forward. Coupling to the efficient electronic friction approach will specifically allow for a statistically meaningful number of trajectories that can better assess the performance of the latter and quantitatively disentangle the contribution of non-adiabatic effects in the presence of a realistic mobile substrate. We further envision corresponding QM/Me+EF studies to establish mechanistic trends for yet more complex systems beyond diatomic adsorbates, while elucidating the potential implications of ‘hot’ chemistry to many important dynamical processes such as the self-assembly of surface nanostructures, the first steps of oxide nucleation and epitaxial growth, or adsorbate-induced surface reconstructions.
Acknowledgements
We gratefully thank Prof. A. C. Luntz for many productive comments and discussions.
Additional information
Funding
Notes
No potential conflict of interest was reported by the authors.
Related Research Data
References
- J.C. Tully, Annu. Rev. Phys. Chem. 31 (1980) p.319–343.
- A. Groß, Theoretical Surface Science: A Microscopic Perspective, 2nd ed., Springer, Heidelberg, 2009.
- J.C. Tully, Annu. Rev. Phys. Chem. 51 (2000) p.153–178.
- A.M. Wodtke, J.C. Tully and D.J. Auerbach, Int. Rev. Phys. Chem. 23 (2004) p.513–539.
- E. Hasselbrink, Curr. Opin. Solid St. M. 10 (2006) p.192–204.
- I. Rahinov, R. Cooper, D. Matsiev, C. Bartels, D.J. Auerbach and A.M. Wodtke, Phys. Chem. Chem. Phys. 13 (2011) p.12680–12692.
- A.M. Wodtke, Chem. Soc. Rev. 45 (2016) p.3641–3657.
- K. Golibrzuch, N. Bartels, D.J. Auerbach and A.M. Wodtke, Annu. Rev. Phys. Chem. 66 (2015) p.399–425.
- I. Chorkendorff and J.W. Niemantsverdriet, Kinetics of reactions on surfaces, chap. 7, Concepts of Modern Catalysis and Kinetics, Wiley-VCH, Weinheim, 2005, pp. 267–299.
- K. Reuter, First-principles kinetic monte carlo simulations for heterogeneous catalysis: Concepts, status and frontiers, in Modeling and Simulation of Heterogeneous Catalytic Reactions: From the Molecular Process to the Technical System, O. Deutschman, ed., Wiley VCH, Weinheim, 2011, pp. 77–111.
- K. Reuter, Catal. Lett. 146 (2016) p.541–563.
- M.K. Sabbe, M.F. Reyniers and K. Reuter, Catal: Sci. Technol. 2 (2012) p.2010–2024.
- K.W. Kolasinski, Surface Science: Foundations of Catalysis and Nanoscience, 3rd ed., John Wiley & Sons, Hoboken, New Jersey, USA, 2012.
- A.C. Luntz, Dynamics of gas-surface interactions, chap. 47, Surface and Interface Science, Wiley-VCH, Weinheim, 2016, pp. 1255–1314.
- A.J. Cohen, P. Mori-Sánchez and W. Yang, Science 321 (2008) p.792–794.
- J.K. Nørskov, F. Abild-Pedersen, F. Studt and T. Bligaard, Proc. Nat. Acad. Sci. USA 108 (2011) p.937–943.
- A. Groß, Surf. Sci. 500 (2002) p.347–367.
- M. Scheffler and C. Stampfl, Theory of adsorption on metal substrates, chap. 5, in Electronic Structure, Handbook of Surface Science, Vol 2, K. Horn and M. Scheffler, eds., Elsevier, Amsterdam, 2000, pp. 285–356.
- A. Groß, Phys. Rev. Lett. 103 (2009) p.246101.
- J.K. Nørskov, T. Bligaard, J. Rossmeisl and C.H. Christensen, Nat. Chem. 1 (2009) p.37–46.
- C.T. Rettner, D.J. Auerbach, J.C. Tully and A.W. Kleyn, J. Phys. Chem. 100 (1996) p.13021–13033.
- L. Vattuone, G. Bracco, M. Smerieri, L. Savio and M. Rocca, Supersonic molecular beams studies of surfaces, chap. 1, in Dynamics of Gas-surface Interactions: Atomic-level Understanding of Scattering Processes at Surfaces, R. Díez Muiño and H.F. Busnengo, eds., Springer, Heidelberg, 2013, pp. 1–23.
- H. Chadwick and R.D. Beck, Chem. Soc. Rev. 45 (2016) p.3576–3594.
- A. Groß and M. Scheffler, Prog. Surf. Sci. 53 (1996) p.187–196.
- A. Groß and M. Scheffler, Phys. Rev. B 57 (1998) p.2493–2506.
- A. Groß, Surf. Sci. Rep. 32 (1998) p.291–340.
- D.A. McCormack, G. Kroes, R.A. Olsen, J.A. Groeneveld, J.N.P. van Stralen, E.J. Baerends and R.C. Mowrey, Faraday Discuss. 117 (2000) p.109–132.
- G.J. Kroes, A. Groß, E.J. Baerends, M. Scheffler and D.A. McCormack, Acc. Chem. Res. 35 (2002) p.193–200.
- C. Díaz, J.K. Vincent, G.P. Krishnamohan, R.A. Olsen, G.J. Kroes, K. Honkala and J.K. Nørskov, Phys. Rev. Lett. 96 (2006) p.096102.
- B. Jiang and H. Guo, J. Chem. Phys. 144 (2016) p.091101.
- X. Shen, Z. Zhang and D.H. Zhang, J. Chem. Phys. 144 (2016) p.101101.
- J. Behler, S. Lorenz and K. Reuter, J. Chem. Phys. 127 (2007) p.014705.
- H.F. Busnengo, A. Salin and W. Dong, J. Chem. Phys. 112 (2000) p.7641–7651.
- V.J. Bukas, J. Meyer, M. Alducin, K. Reuter, Z. Phys. Chem. 227 (2013) p.1523–1542.
- A. Groß, Surf. Sci. 606 (2012) p.690–691.
- A. Groß, Appl. Phys. A 67 (1998) p.627–635.
- G.J. Kroes and C. Díaz, Chem. Soc. Rev. 45 (2016) p.3658.
- G.J. Kroes, Science 321 (2008) p.794.
- C. Díaz, E. Pijper, R.A. Olsen, H.F. Busnengo, D.J. Auerbach and G.J. Kroes, Science 326 (2009) p.832.
- H. Guo, A. Farjamnia and B. Jackson, J. Phys. Chem. Lett. 7 (2016) p.4576–4584.
- B. Jiang, M. Yang, D. Xie and H. Guo, Chem. Soc. Rev. 45 (2016) p.3621–3640.
- J. Behler and M. Parrinello, Phys. Rev. Lett. 98 (2007) p.146401.
- J. Behler, J. Chem. Phys. 134 (2011) p.074106.
- J. Behler, J. Chem. Phys. 145 (2016) p.170901.
- B. Kolb, X. Luo, X. Zhou, B. Jiang and H. Guo, J. Phys. Chem. Lett. 8 (2017) p.666–672.
- K. Shakouri, J. Behler, J. Meyer and G.J. Kroes, J. Phys. Chem. Lett. 8 (2017) p.2131–2136.
- M. Hand and J. Harris, J. Chem. Phys. 92 (1990) p.7610–7617.
- A. Luntz and J. Harris, Surf. Sci. 258 (1991) p.397–426.
- S.A. Adelman and J.D. Doll, J. Chem. Phys. 64 (1976) p.2375–2388.
- M. Shugard, J.C. Tully and A. Nitzan, J. Chem. Phys. 66 (1977) p.2534–2544.
- J.C. Tully, J. Chem. Phys. 73 (1980) p.1975–1985.
- J.C. Polanyi and R.J. Wolf, J. Chem. Phys. 82 (1985) p.1555–1566.
- M. Dohle and P. Saalfrank, Surf. Sci. 373 (1997) p.95–108.
- H.F. Busnengo, W. Dong, P. Sautet and A. Salin, Phys. Rev. Lett. 87 (2001) p.127601.
- E. Watts and G.O. Sitz, J. Chem. Phys. 111 (1999) p.9791–9796.
- Z.S. Wang, G.R. Darling and S. Holloway, Phys. Rev. Lett. 87 (2001) p.226102.
- H.F. Busnengo, W. Dong and A. Salin, Phys. Rev. Lett. 93 (2004) p.236103.
- H.F. Busnengo, M.A. Di Césare, W. Dong and A. Salin, Phys. Rev. B 72 (2005) p.125411.
- V.J. Bukas, S. Mitra, J. Meyer and K. Reuter, J. Chem. Phys. 143 (2015) p.034705.
- A. den Dunnen, S. Wiegman, L. Jacobse and L.B.F. Juurlink, J. Chem. Phys. 142 (2015) p.214708.
- L. Martin-Gondre, M. Alducin, G.A. Bocan and R. Díez Muiño, and J.I. Juaristi, Phys. Rev. Lett. 108 (2012) p.096101.
- L. Martin-Gondre, M. Alducin, G.A. Bocan and R. Díez Muiño, and J.I. Juarist, Phys. Rev. Lett. 108 (2012) p.139901(E).
- M. Blanco-Rey, E. Díaz, G.A. Bocan and R. Díez Muiño, M. Alducin, and J.I. Juaristi, J. Phys. Chem. Lett. 4 (2013) p.3704–3709.
- R. Pétuya, P.A. Plötz, C. Crespos and P. Larrégaray, J. Phys. Chem. C 118 (2014) p.21904–21910.
- I. Goikoetxea, J. Meyer, J.I. Juaristi, M. Alducin and K. Reuter, Phys. Rev. Lett. 112 (2014) p.156101.
- J.P. Toennies, J. Phys. Condens. Matter 5 (1993) p.A25.
- M.D. Stiles and J.W. Wilkins, Phys. Rev. Lett. 54 (1985) p.595–598.
- M.D. Stiles, J.W. Wilkins and M. Persson, Phys. Rev. B 34 (1986) p.4490–4510.
- B. Jackson, Comput. Phys. Commun. 80 (1994) p.119–144.
- M. Dohle, P. Saalfrank and T. Uzer, Surf. Sci. 409 (1998) p.37–45.
- M. Dohle, P. Saalfrank and T. Uzer, J. Chem. Phys. 108 (1998) p.4226–4236.
- D.J. Auerbach, Phys. Scri. 1983 (1983) p.122.
- A.S. Sanz and S. Miret-Artés, Phys. Rep. 451 (2007) p.37–154.
- C. Carbogno, A. Groß, J. Meyer and K. Reuter, Dynamics of gas-surface interactions: Atomic-level understanding of scattering processes at surfaces, chap. 16, in Dynamics of Gas-surface Interactions: Atomic-level Understanding of Scattering Processes at Surfaces, R. Díez Muiño and F.H. Busnengo, eds., Springer, Heidelberg, 2013, pp. 389–419.
- V.J. Bukas and K. Reuter, J. Chem. Phys. 146 (2017) p.014702.
- A.C. Luntz and R.D. Beck, J. Vac. Sci. Technol. A 35 (2017) p.05C201.
- P.M. Hundt, B. Jiang, M.E. van Reijzen, H. Guo and R.D. Beck, Science 344 (2014) p.504–507.
- X. Zhou, B. Kolb, X. Luo, H. Guo and B. Jiang, J. Phys. Chem. C 121 (2017) p.5594–5602.
- A.K. Tiwari, S. Nave and B. Jackson, Phys. Rev. Lett. 103 (2009) p.253201.
- A.K. Tiwari, S. Nave and B. Jackson, J. Chem. Phys. 132 (2010) p.134702.
- B. Jackson, The effects of lattice motion on gas-surface reactions, chap. 9, in Dynamics of Gas-Surface Interactions: Atomic-level Understanding of Scattering Processes at Surfaces, R. Díez Muiño and H.F. Busnengo, eds., Springer, Heidelberg, 2013, pp. 213–237.
- B. Jackson and S. Nave, J. Chem. Phys. 138 (2013) p.174705.
- A. Groß, Dynamics of reactions at surfaces, chap. 2, in Modeling and Simulation of Heterogeneous Catalytic Reactions, Wiley-VCH, Weinheim, 2011, pp. 39–70.
- F. Nattino, H. Ueta, H. Chadwick, M.E. van Reijzen, R.D. Beck, B. Jackson, M.C. van Hemert and G.J. Kroes, J. Phys. Chem. Lett. 5 (2014) p.1294–1299.
- F. Nattino, C. Díaz, B. Jackson and G.J. Kroes, Phys. Rev. Lett. 108 (2012) p.236104.
- G. Füchsel, M. del Cueto, C. Díaz and G.J. Kroes, J. Phys. Chem. C 120 (2016) p.25760–25779.
- F. Nattino, F. Costanzo and G.J. Kroes, J. Chem. Phys. 142 (2015) p.104702.
- F. Nattino, D. Migliorini, M. Bonfanti and G.J. Kroes, J. Chem. Phys. 144 (2016) p.044702.
- A. Groß, Catal. Today 260 (2016) p.60–65.
- F. Nattino, O. Galparsoro, F. Costanzo and R. Díez Muiño, M. Alducin, and G.J. Kroes, J. Chem. Phys. 144 (2016) p.244708.
- P.R. Shirhatti, J. Geweke, C. Steinsiek, C. Bartels, I. Rahinov, D.J. Auerbach and A.M. Wodtke, J. Phys. Chem. Lett. 7 (2016) p.1346–1350.
- B. Kolb and H. Guo, J. Chem. Phys. 145 (2016) p.011102.
- J. Behler, B. Delley, S. Lorenz, K. Reuter and M. Scheffler, Phys. Rev. Lett. 94 (2005) p.036104.
- C. Carbogno, J. Behler, A. Groß and K. Reuter, Phys. Rev. Lett. 101 (2008) p.096104.
- F. Libisch, C. Huang, P. Liao, M. Pavone and E.A. Carter, Phys. Rev. Lett. 109 (2012) p.198303.
- Y. Huang, A.M. Wodtke, H. Hou, C.T. Rettner and D.J. Auerbach, Phys. Rev. Lett. 84 (2000) p.2985–2988.
- Y. Huang, C.T. Rettner, D.J. Auerbach and A.M. Wodtke, Science 290 (2000) p.111.
- S. Li and H. Guo, J. Chem. Phys. 117 (2002) p.4499–4508.
- J.C. Tully, Faraday Discuss. 110 (1998) p.407–419.
- J.C. Tully, Nonadiabatic Dynamics, chap. 2, in Modern Methods for Multidimensional Dynamics Computations in Chemistry, D.L. Thompson, ed., World Scientific, Singapore, 1998, pp. 34–72.
- M. Lindenblatt and E. Pehlke, Phys. Rev. Lett. 97 (2006) p.216101.
- M. Grotemeyer and E. Pehlke, Phys. Rev. Lett. 112 (2014) p.043201.
- J.C. Tully, J. Chem. Phys. 93 (1990) p.1061–1071.
- M.D. Hack and D.G. Truhlar, J. Phys. Chem. A 104 (2000) p.7917–7926.
- N.A. Shenvi, S. Roy and J.C. Tully, J. Chem. Phys. 130 (2009) p.174107.
- W. Dou and J.E. Subotnik, J. Chem. Theo. Comp. 13 (2017) p.2430–2439.
- N.A. Shenvi, S. Roy and J.C. Tully, Science 326 (2009) p.829–832.
- S. Roy, N.A. Shenvi and J.C. Tully, J. Chem. Phys. 130 (2009) p.174716.
- M. Timmer and P. Kratzer, Phys. Rev. B 79 (2009) p.165407.
- J. Meyer and K. Reuter, New J. Phys. 13 (2011) p.085010.
- S. Rittmeyer, J. Meyer and K. Reuter, Phys. Rev. Lett. 119 (2017) p.176808. doi:10.1103/PhysRevLett.119.176808.
- E.G. d’Agliano, P. Kumar, W. Schaich and H. Suhl, Phys. Rev. B 11 (1975) p.2122–2143.
- Y. Li and G. Wahnström, Phys. Rev. Lett. 68 (1992) p.3444–3447.
- M. Head-Gordon and J.C. Tully, J. Chem. Phys. 103 (1995) p.10137–10145.
- W. Dou, A. Nitzan and J.E. Subotnik, J. Chem. Phys. 143 (2015) p.054103.
- W. Dou and J.E. Subotnik, J. Chem. Phys. 146 (2017) p.092304.
- W. Dou, G. Miao and J.E. Subotnik, Phys. Rev. Lett. 119 (2017) p.046001.
- T.L. Ferrell and R.H. Ritchie, Phys. Rev. B 16 (1977) p.115–123.
- P. Echenique, R. Nieminen and R. Ritchie, Solid State Commun. 37 (1981) p.779–781.
- P. Sigmund, Phys. Rev. A 26 (1982) p.2497–2517.
- K. Schönhammer and O. Gunnarsson, Energy dissipation at metal surfaces: The electron hole-pair mechanism, in Many-Body Phenomena at Surfaces, D. Langreth and H. Suhl, eds., Academic Press, Orlando, Florida, 1984, pp. 421–451.
- B. Hellsing and M. Persson, Phys. Scr. 29 (1984) p.360.
- J.I. Juaristi, M. Alducin and R. Díez Muiño, H.F. Busnengo, and A. Salin, Phys. Rev. Lett. 100 (2008) p.116102.
- M.J. Puska and R.M. Nieminen, Phys. Rev. B 27 (1983) p.6121–6128.
- P.M. Echenique, R.M. Nieminen, J.C. Ashley and R.H. Ritchie, Phys. Rev. A 33 (1986) p.897–904.
- P. Echenique and M. Uranga, Density functional theory of stopping power, in Interaction of Charged Particles with Solids and Surfaces, Nato ASI Series, Vol. 271, A. Gras-Marti, H.M. Urbassek, N.R. Arista and F. Flores, eds., Plenum Press, New York, 1991, pp. 39–71.
- M. Askerka, R.J. Maurer, V.S. Batista and J.C. Tully, Phys. Rev. Lett. 116 (2016) p.217601.
- R.J. Maurer, M. Askerka, V.S. Batista and J.C. Tully, Phys. Rev. B 94 (2016) p.115432.
- J. Trail, M. Graham and D. Bird, Comp. Phys. Comm. 137 (2001) p.163–173.
- N. Lorente and M. Persson, Faraday Discuss. 117 (2000) p.277–290.
- R.J. Maurer, B. Jiang, H. Guo and J.C. Tully, Phys. Rev. Lett. 118 (2017) p.256001.
- J.R. Trail, M.C. Graham, D.M. Bird, M. Persson and S. Holloway, Phys. Rev. Lett. 88 (2002) p.166802.
- J.R. Trail, D.M. Bird, M. Persson and S. Holloway, J. Chem. Phys. 119 (2003) p.4539–4549.
- A.C. Luntz and M. Persson, J. Chem. Phys. 123 (2005) p.074704.
- A.C. Luntz, M. Persson, S. Wagner, C. Frischkorn and M. Wolf, J. Chem. Phys. 124 (2006) p.244702.
- A.C. Luntz, M. Persson and G.O. Sitz, J. Chem. Phys. 124 (2006) p.091101.
- G. Füchsel, T. Klamroth, S. Monturet and P. Saalfrank, Phys. Chem. Chem. Phys. 13 (2011) p.8659–8670.
- A.C. Luntz, I. Makkonen, M. Persson, S. Holloway, D.M. Bird and M.S. Mizielinski, Phys. Rev. Lett. 102 (2009) p.109601.
- J.I. Juaristi, M. Alducin and R. Díez Muiño, H.F. Busnengo, and A. Salin, Phys. Rev. Lett. 102 (2009) p.109602.
- F. Hirshfeld, Theor. Chim. Acta 44 (1977) p.129–138.
- S.P. Rittmeyer, J. Meyer, J.I. Juaristi and K. Reuter, Phys. Rev. Lett. 115 (2015) p.046102.
- I. Goikoetxea, J.I. Juaristi, M. Alducin and R. Díez Muiño, J. Phys. Condens. Matter 21 (2009) p.264007.
- B. Jiang, M. Alducin and H. Guo, J. Phys. Chem. Lett. 7 (2016) p.327–331.
- X. Luo, B. Jiang, J.I. Juaristi, M. Alducin and H. Guo, J. Chem. Phys. 145 (2016) p.044704.
- A.S. Muzas, J.I. Juaristi, M. Alducin and R. Díez Muiño, G.J. Kroes, and C. Díaz, J. Chem. Phys. 137 (2012) p.064707.
- G. Füchsel, S. Schimka and P. Saalfrank, J. Phys. Chem. A 117 (2013) p.8761–8769.
- M. Head-Gordon and J.C. Tully, Phys. Rev. B 46 (1992) p.1853–1856.
- V. Krishna and J.C. Tully, J. Chem. Phys. 125 (2006) p.054706.
- M. Forsblom and M. Persson, J. Chem. Phys. 127 (2007) p.154303.
- H. Arnolds, Prog. Surf. Sci. 86 (2011) p.1–40.
- R. Cooper, C. Bartels, A. Kandratsenka, I. Rahinov, N. Shenvi, K. Golibrzuch, Z. Li, D.J. Auerbach, J.C. Tully and A.M. Wodtke, Angew. Chem. Int. Ed. 51 (2012) p.4954–4958.
- J.T. Kindt, J.C. Tully, M. Head-Gordon and M.A. Gomez, J. Chem. Phys. 109 (1998) p.3629–3636.
- L. Martin-Gondre, G. Bocan, M. Alducin, J. Juaristi and R. Díez Muiño, Comput. Theor. Chem. 990 (2012) p.126–131.
- L. Martin-Gondre, G.A. Bocan, M. Blanco-Rey, M. Alducin, J.I. Juaristi and R. Díez Muiño, J. Phys. Chem. C 117 (2013) p.9779–9790.
- G.J. Kroes, M. Pavanello, M. Blanco-Rey, M. Alducin and D.J. Auerbach, J. Chem. Phys. 141 (2014) p.054705.
- S.M. Janke, M. Pavanello, G.J. Kroes, D. Auerbach, A.M. Wodtke, A. Kandratsenka, Z. Phys. Chem. 227 (2013) p.1467–1490.
- O. Bünermann, H. Jiang, Y. Dorenkamp, A. Kandratsenka, S.M. Janke, D.J. Auerbach and A.M. Wodtke, Science 350 (2015) p.1346–1349.
- S.M. Janke, D.J. Auerbach, A.M. Wodtke and A. Kandratsenka, J. Chem. Phys. 143 (2015) p.124708.
- O. Galparsoro, R. Pétuya, J.I. Juaristi, C. Crespos, M. Alducin and P. Larrégaray, J. Phys. Chem. C 119 (2015) p.15434–15442.
- O. Galparsoro, J.I. Juaristi, C. Crespos, M. Alducin and P. Larrégaray, J. Phys. Chem. C (2017).
- P. Saalfrank, Chem. Rev. 106 (2006) p.4116–4159.
- B.N.J. Persson, J. Phys. C: Solid State Phys. 11 (1978) p.4251.
- B. Persson and M. Persson, Surf. Sci. 97 (1980) p.609–624.
- B. Persson and M. Persson, Solid State Commun. 36 (1980) p.175–179.
- M. Persson and B. Hellsing, Phys. Rev. Lett. 49 (1982) p.662–665.
- B. Hellsing, M. Persson and B. Lundqvist, Surf. Sci. 126 (1983) p.147–153.
- B.N.J. Persson, J. Phys. C: Solid State Phys. 17 (1984) p.4741.
- J.C. Tully, M. Gomez and M. Head-Gordon, J. Vac. Sci. Technol. A 11 (1993) p.1914–1920.
- M. Morin, N.J. Levinos and A.L. Harris, J. Chem. Phys. 96 (1992) p.3950–3956.
- P. Saalfrank, J.I. Juaristi, M. Alducin, M. Blanco-Rey and R. Díez Muiño, J. Chem. Phys. 141 (2014) p.234702.
- M. Head-Gordon and J.C. Tully, J. Chem. Phys. 96 (1992) p.3939–3949.
- M.S. Mizielinski, D.M. Bird, M. Persson and S. Holloway, J. Chem. Phys. 122 (2005) p.084710.
- J. Barth, Surf. Sci. Rep. 40 (2000) p.75–149.
- T. Ala-Nissila, R. Ferrando and S.C. Ying, Adv. Phys. 51 (2002) p.949–1078.
- G. Wahnström, A.B. Lee and J. Strömquist, J. Chem. Phys. 105 (1996) p.326–336.
- S.P. Rittmeyer, D.J. Ward, P. Gütlein, J. Ellis, W. Allison and K. Reuter, Phys. Rev. Lett. 117 (2016) p.196001.
- A. Jardine, H. Hedgeland, G. Alexandrowicz, W. Allison and J. Ellis, Prog. Surf. Sci. 84 (2009) p.323–379.
- A.P. Jardine, J. Ellis and W. Allison, J. Chem. Phys. 120 (2004) p.8724–8733.
- A.P. Jardine, G. Alexandrowicz, H. Hedgeland, W. Allison and J. Ellis, Phys. Chem. Chem. Phys. 11 (2009) p.3355–3374.
- A. Carley, P. Davies and M. Roberts, Catal. Lett. 80 (2002) p.25–34.
- H. Brune, J. Wintterlin, R.J. Behm and G. Ertl, Phys. Rev. Lett. 68 (1992) p.624–626.
- J. Wintterlin, R. Schuster and G. Ertl, Phys. Rev. Lett. 77 (1996) p.123–126.
- B.G. Briner, M. Doering, H.P. Rust and A.M. Bradshaw, Phys. Rev. Lett. 78 (1997) p.1516–1519.
- S. Schintke, S. Messerli, K. Morgenstern, J. Nieminen and W.D. Schneider, J. Chem. Phys. 114 (2001) p.4206–4209.
- M. Rose, A. Borg, J. Dunphy, T. Mitsui, D. Ogletree and M. Salmeron, Surf. Sci. 561 (2004) p.69–78.
- M.F. Hsieh, D.S. Lin, H. Gawronski and K. Morgenstern, J. Chem. Phys. 131 (2009) p.174709.
- C. Sprodowski, M. Mehlhorn and K. Morgenstern, Z. Phys. Chem. 22 (2010) p.264005.
- M. Schmid, G. Leonardelli, R. Tscheließnig, A. Biedermann and P. Varga, Surf. Sci. 478 (2001) p.L355–L362.
- A.J. Komrowski, J.Z. Sexton, A.C. Kummel, M. Binetti, O. Weiße and E. Hasselbrink, Phys. Rev. Lett. 87 (2001) p.246103.
- V.J. Bukas and K. Reuter, Phys. Rev. Lett. 117 (2016) p.146101.
- O. Galparsoro, R. Petuya, F. Busnengo, J.I. Juaristi, C. Crespos, M. Alducin and P. Larrégaray, Phys. Chem. Chem. Phys. 18 (2016) p.31378–31383.
- C. Engdahl and G. Wahnström, Surf. Sci. 312 (1994) p.429–440.
- M. Blanco-Rey, J.I. Juaristi and R. Díez Muiño, H.F. Busnengo, G.J. Kroes, and M. Alducin, Phys. Rev. Lett. 112 (2014) p.103203.
- D. Novko, M. Blanco-Rey, J.I. Juaristi and M. Alducin, Phys. Rev. B 92 (2015) p.201411.
- D. Novko, M. Blanco-Rey, J. Juaristi and M. Alducin, Nucl. Instrum. Meth. B 382 (2016) p.26–31.
- P. Nieto, E. Pijper, D. Barredo, G. Laurent, R.A. Olsen, E.J. Baerends, G.J. Kroes and D. Farías, Science 312 (2006) p.86–89.
- J. Meyer and K. Reuter, Angew. Chem. Int. Ed. 53 (2014) p.4721–4724.
- M.S. Daw and M.I. Baskes, Phys. Rev. B 29 (1984) p.6443–6453.
- M.I. Baskes, Phys. Rev. B 46 (1992) p.2727–2742.
- B. Jackson, Comput. Phys. Commun. 63 (1991) p.154–170.
- B. Jackson, J. Chem. Phys. 108 (1998) p.1131–1139.