
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Despite the key role that ionic solutions play in several natural and industrial processes, a unified, molecular-level understanding of how ions affect the structure and dynamics of water across different phases remains elusive. In this context, computer simulations can provide new insights that are difficult, if not impossible, to obtain by other means. However, the predictive power of a computer simulation directly depends on the level of ‘realism’ that is used to represent the underlying molecular interactions. Here, we report a systematic analysis of many-body effects in halide-water clusters and demonstrate that the recently developed MB-nrg full-dimensional many-body potential energy functions achieve high accuracy by quantitatively reproducing the individual terms of the many-body expansion of the interaction energy, thus opening the door to realistic computer simulations of ionic solutions.
Graphical Abstract
I. Introduction
A quantitative characterization of the driving forces and molecular mechanisms that regulate ion hydration is key to the microscopic understanding of fundamental processes taking place in aqueous clusters, solutions, and interfaces, which, in turn, have major implications for different fields of science and engineering. For example, charged species are often found as intermediates in chemical reactions and catalytic processes [Citation1,Citation2]. In biochemistry, ions play a central role in the stabilization of biomolecules [Citation3–Citation5] as well as in mediating protein-protein interactions [Citation6,Citation7], intracellular signal transduction [Citation8,Citation9], and enzyme and nucleic acid catalysis [Citation10–Citation13]. In the atmosphere, ionic clusters carry electric currents and are involved in the formation and evolution of aerosol particles [Citation14,Citation15]. In materials science, ionic solutions are central to many devices such as electrolytic cells, capacitors, and batteries [Citation16].
Due to the long-range nature of Coulomb interactions, ions do not exist as isolated species under equilibrium at ambient conditions in the gas phase, but form neutral ionic aggregates (e.g. ionic salts or, in special cases, ionic liquids). Completely different behavior is observed when ions are dissolved in water. In this case, ion-ion interactions are mediated by the presence of water molecules that allow ions to exist as individual-solvated species, up to saturation. The stabilization of individual-hydrated ions results from the interplay of energetic contributions associated with ion-water interactions, which counteract the direct Coulomb attraction between ions of opposite charges, and entropic contributions associated with the reorganization of the water hydrogen-bond (H-bond) network required to accommodate the ionic species. Given the central role played by water in mediating many fundamental processes, it is not surprising that much experimental and theoretical work has been and continues to be devoted to the development of a microscopic understanding of ion hydration [Citation17–Citation20].
The first attempt to rationalize specific ion effects on the properties of aqueous solutions dates back to Hofmeister who classified different salts according to their ability to precipitate egg-white proteins and, subsequently, explained his observations in terms of the corresponding strength of hydration [Citation21,Citation22]. Over the years, Hofmeister’s original explanation has led to the development of the concept of ‘structure makers’ and ‘structure breakers’ to indicate ions that are strongly and weakly hydrated, respectively [Citation23,Citation24]. Nearly equivalent terms ‘kosmotropes’ and ‘chaotropes’ were later introduced in biophysical contexts to identify ‘structure makers’ and ‘structure breakers’, respectively [Citation25]. Within this picture, small and highly charged ions are considered as ‘structure makers’, strengthening the water H-bond network and inducing tetrahedral order. In contrast, large, monovalent ions correspond to ‘structure breakers’, disrupting the water H-bond network and leading to a more disordered liquid structure. This classification, which finds support in measurements of macroscopic properties (e.g. viscosity and ion mobility), implies that ions that act as ‘structure makers’ can affect the structural properties of aqueous solutions at long range, effectively inducing long-range ordering.
Measurements carried out using different vibrational spectroscopic techniques, including terahertz [Citation26,Citation27], infrared [Citation18], and Raman [Citation28] spectroscopies as well as X-ray absorption spectroscopy [Citation29,Citation30], have challenged this picture, suggesting that dissolved ions have no influence on the water H-bond network at long range. However, this interpretation is apparently at odds with infrared photodissociation spectra measured for sulfate ions in water clusters, which indicate that sulfate ions induce H-bond ordering beyond the first hydration shell [Citation31]. Femtosecond elastic second harmonic scattering (fs-ESHS) measurements carried out for highly dilute (from 10 μM to few mM concentration) salt solutions have provided further support for long-range ion-induced structural ordering by detecting orientational correlations between water molecules at distances up to 10 nm from the ions, which were originally explained invoking ion-dipole and collective H-bond interactions [Citation32]. Alternative interpretations of these apparent long-range effects have recently been proposed from molecular dynamics (MD) simulations [Citation33,Citation34].
One of the most striking examples of present uncertainties associated with our understanding of specific ion effects is perhaps represented by the debate around the distribution of ions at the air/water interface [Citation17,Citation20]. Measurements carried out by Heydweiller more than 100 years ago showed that the surface tension of electrolyte solutions is larger than that of pure water, and displays a strong dependence on the nature of the anions but varies little as a function of the cations [Citation35]. This trend, which follows a reverse Hofmeister series, was independently explained by Wagner [Citation36], and Onsager and Samaras [Citation37] by considering the effects of image charges which cause a depletion of ions in the interfacial region and thus account for the observed increase in surface tension. Subsequent experiments, however, found that the electrostatic potential difference across the air/water interface measured for solutions of halide salts (with the exception of fluoride salts) is more negative than that for pure water, thus suggesting that larger halide ions have higher propensity for the interface than cations, in contradiction with predictions derived from Wagner, and Onsager and Samaras theories [Citation38].
Renewed interest in specific ion effects at aqueous interfaces was generated by the development of both surface-sensitive experimental techniques. In particular, vibrational sum-frequency generation (vSFG) spectra measured for NaF and NaCl solutions were found to be very similar to those of neat water while appreciable differences were found in the analogous spectra of NaBr and NaI solutions [Citation39]. Since the vSFG spectra are sensitive to changes in the water H-bond network, the results of Ref. [Citation39] were interpreted as an indication of the presence of Br- and I-ions in the interfacial region. Opposite conclusions were, however, reached from analogous vSFG studies in Ref. [Citation40], which indicated ‘a significantly diminished population of the anions at the uppermost layer of the surface region.’ Subsequent analyses of second harmonic generation (SHG) spectra in terms of Langmuir adsorption isotherms as well as photoelectron (PE) measurements of anion/cation ratios as a function of photoelectron kinetic energy provided further support for the adsorption of larger halide ions at the air/water interface [Citation41].
On the theoretical side, pioneering MD simulations of halide-water clusters, carried out with polarizable force fields (FFs), found that all halide ions except fluoride had a preference for the surface of the clusters [Citation42–Citation45]. Analogous simulations carried out with nonpolarizable FFs predicted anions to always be in the interior of the clusters, thus emphasizing the importance of many-body effects in ion-water interactions [Citation46–Citation48]. Subsequent MD simulations of concentrated electrolyte solutions as well as calculations of single-ion potentials of mean force (PMFs) carried out in slab geometries using polarizable FFs found a significant adsorption of the larger halide ions at the interface, with the surface propensity increasing from Cl – to I – while F – as well as Na+ ions were repelled from the interface [Citation49,Citation50]. Qualitatively similar predictions were made from analogous MD simulations carried out with different polarizable models [Citation51–Citation53]. In an attempt to reconcile Wagner, and Onsager and Samaras theory with recent surface-sensitive measurements and computer simulations, an extension of the dielectric continuum (DC) theory has been proposed which takes into account both the dimension and the polarizability of the ions [Citation54]. Within the extended DC theory, it is predicted that highly polarizable ions are indeed adsorbed at the water surface, although to a much less extent than found in MD simulations with polarizable FFs. An alternative explanation, emphasizing the impact that ions may have on surface fluctuations, has also been proposed [Citation55]. More recently, MD simulations based on density functional theory (DFT) reported a much lower propensity of iodide for the water surface compared to predictions obtained with polarizable FFs [Citation56]. However, no further simulations for other ions have so far been reported to provide support for similar differences between polarizable FFs and DFT models.
While much progress has been made over the last two decades in identifying different factors that affect ion hydration in different environments, it has become increasingly apparent that any further step towards a unified and quantitative theory of ion hydration requires an accurate description of the underlying molecular interactions. In this article, we systematically investigate many-body effects in halide-water interactions and provide a quantitative assessment of the accuracy of recently developed many-body (MB) potential energy functions (PEFs) in comparison with polarizable FFs and various DFT models that are commonly employed in computer simulations of electrolyte solutions. With this objective in mind, in Section 2 we introduce the many-body expansion (MBE) of the interaction energy and provide a brief historical perspective on its application to aqueous systems. Section 3 briefly overviews recently developed MB PEFs. A systematic analysis of many-body effects in halide hydration is presented in Section 4 along with detailed comparisons between MB PEFs, polarizable FFs, and various DFT models. Conclusions and outlook are given in Section 5.
2. Many-body expansion of the interaction energy
The energy of a system containing N (atomic or molecular) monomers can be formally expressed through the MBE as [Citation57]
where ri collectively denotes the coordinates of all atoms within the ith monomer. In Eq. (1), V1B is the one-body (1B) potential representing the energy required to deform an individual monomer from its equilibrium geometry. Therefore, by construction, V1B(i) = 0 for atomic monomers, and V1B(i) = E(i) – Eeq(i) for molecular monomers, where E(i) and Eeq(i) are the monomer energies for distorted and equilibrium configurations, respectively. All higher n-body (nB) interaction terms (VnB) in Eq. (1) are defined recursively through
The importance of many-body effects in water-water and ion-water interactions was already recognized in the 1950s by Frank and Wen who introduced a molecular picture of water consisting of ‘flickering clusters of hydrogen-bonded molecules’, emphasizing the ‘cooperative nature’ of hydrogen bonding [Citation58]. Pioneering electronic structure calculations carried out on small aqueous clusters demonstrated that nonadditive contributions to the interaction energy beyond the pairwise additive 2B term are not negligible [Citation59,Citation60]. In particular, it was found that the 3B term can contribute up to ∼15–20% of the interaction energy of cyclic water structures, while 4B contributions represent, on average, ∼1% of the interaction energy in generic water clusters [Citation61–Citation71].
Due to the rapid convergence of the MBE for non-metallic systems, Eq. 1 also provides a rigorous and efficient framework for the development of full-dimensional PEFs in which individual terms of the MBE can be determined using different electronic structure methods. Most empirical FFs describing water-water and ion-water interactions are pairwise additive (i.e. the MBE is truncated at the 2B term) and attempt to account for many-body contributions in an average fashion by using an effective V2B [Citation72–Citation83]. Although in the early times of MD simulations this simplification was a necessity dictated by computational efficiency, it was soon recognized that ‘pair potentials do not realistically reproduce both gas and condensed phase water properties’ [Citation84]. The first attempts to derive PEFs for aqueous systems which could rigorously represent the individual terms of the MBE were made in the late 1970s and 1980s [Citation85–Citation90]. In particular, Clementi and coworkers developed a series of analytical PEFs for water which were fitted to ab initio reference data obtained at the fourth-order Møller-Plesset (MP4) and Hartree-Fock levels of theory for the 2B and 3B terms, respectively, and represented many-body effects implicitly through a classical polarization term [Citation89,Citation90]. Stillinger and David developed a polarizable model for water in which treats H+ and O2- were considered as the basic dynamical and structural elements [Citation91]. Building upon these pioneering studies, several polarizable FFs for water have been developed over the years (see Ref. [Citation92] for a recent review).
Nonadditive effects in ion-water interactions were first investigated by Kollman and co-workers who developed a series of polarizable FFs specifically parameterized to reproduce several experimental data [Citation84]. These polarizable FFs were subsequently used in MD simulations of small Cl−(H2O)n and Na+(H2O)n clusters, which provided the first indication of the propensity of polarizable halide ions for the water surface [Citation42–Citation45]. Several empirical polarizable FFs with different degrees of sophistication were then developed and shown to reproduce, at least to some extent, several properties of ion-water systems, from small clusters to bulk solutions and interfaces [Citation46–Citation48,Citation93]. Although these studies emphasized the importance of polarization in correctly representing ion-water interactions, especially in the case of large anions, it was shown that predictions of MD simulations carried out with polarizable FFs depend sensitively on the type of electrostatic dampening adopted in the models, which can often lead to overpolarization of the ions at short ion-water distances [Citation94–Citation96].
The development of efficient algorithms for correlated electronic structure methods along with continued improvements in computer performance has recently made possible to determine, with chemical accuracy, individual (low-order) terms of Eq. 1. In parallel, tremendous progress has been made in constructing multidimensional mathematical functions that are capable to reproduce interaction energies in generic N-molecule systems, with high fidelity [Citation97–Citation99]. By combining these three factors, it has been realized that the MBE effectively provides a rigorous and efficient framework for the development of full-dimensional PEFs entirely from ‘first principles’, in which the low-order terms are accurately determined from correlated electronic structure data (e.g. using coupled cluster theory with single, double, and perturbative triple excitations, CCSD(T), in the complete basis set, CBS, limit, the current ‘gold standard’ for chemical accuracy) and the higher-order terms are represented implicitly by classical many-body induction. Along these lines, several MB PEFs for water have been proposed in the last decade, the most notable of which are CC-pol [Citation100], WHBB [Citation101], HBB2-pol [Citation102], and MB-pol [Citation103–Citation105]. When employed in computer simulations that allow for explicit treatment of nuclear quantum effects, these many-body PEFs have been shown to correctly predict structural, thermodynamic, dynamical, and spectroscopic properties of water, from the dimer in the gas phase to liquid water and ice (see Ref. [Citation92] for a recent review). Among existing many-body PEFs for water, MB-pol has been shown to correctly predict the vibration-rotation tunneling spectrum of the water dimer [Citation103], the energetics, quantum equilibria, and infrared spectra of small clusters [Citation104,Citation106–Citation108], the structural, thermodynamic, and dynamical properties of liquid water [Citation105,Citation109], the energetics of different ice phases [Citation110], infrared and Raman spectra of liquid water [Citation111–Citation113], the vibrational sum-frequency generation spectrum of the air/water interface [Citation114,Citation115], the infrared and Raman spectra of ice Ih [Citation116]. More recently, molecular configurations extracted from classical (MD) and quantum path-integral molecular dynamics (PIMD) simulations with MB-pol have been used to determine the electronic band gap of liquid water, both in the bulk and at the air/water interface, through many-body perturbation theory electronic structure calculations [Citation117].
3. Many-body potential energy functions for halide-water interactions
Building upon the accuracy and computational efficiency demonstrated by MB-pol in predicting the properties of water across different phases [Citation118], the MBE in Eq. 1 has recently been applied to the development of two families of MB PEFs (TTM-nrg for ‘Thole-type model energy’ [Citation119] and MB-nrg for ‘many-body energy’ [Citation120]) describing halide-water interactions. Both TTM-nrg and MB-nrg PEFs adopt MB-pol to describe all water properties (i.e. water monomer distortion, dipole moment, and polarizability, as well as water-water interactions) [Citation118] and have been derived from CCSD(T)-F12b/CBS data for halide-water dimers and halide-water-water trimers but differ in the functional forms employed to represent halide-water many-body effects.
In the TTM-nrg PEFs, halide-water interactions are described by four terms,
where represents permanent and induced electrostatic interactions, with the induction term being derived from the extended Thole-type model originally introduced in Ref. [Citation121]. The repulsive term,
, is represented by a sum of pairwise Born-Mayer functions
between the halide ion (X) and the oxygen (O) and hydrogen (H1 and H2) atoms of the ith water molecule in the system,
where are interatomic distances, and
and
are fitting parameters, with Y = O, H1, and H2. Similarly, the dispersion energy,
, is also represented through a sum of pairwise additive contributions,
,
where are Tang-Toennies damping functions [Citation122] with coefficients
, and
are dispersion coefficients calculated using the exchange-hole dipole model (XDM) [Citation123].
In the MB-nrg PEFs, the interactions between individual halide ions and water molecules are described through the MBE of Eq. 1 and include explicit 2B (ion-water) and 3B (ion-water-water) terms, with all higher-order interactions being implicitly taken into account through many-body induction as in the TTM-nrg model. The 2B term of the MB-nrg PEFs includes three contributions,
where the electrostatic term corresponds to the 2B component of
in Eq. 3 and
is the same sum of pairwise additive contributions as in the TTM-nrg model (Eq. 5).
is expressed by a 4th-degree permutationally invariant polynomial [Citation97] in variables that are functions of the distances between the ion and the six sites of an MB-pol water molecule. Similarly, the 3B term of the MB-nrg PEFs includes two contributions,
where the electrostatic term corresponds to the 3B component of
in Eq. 3 and
is expressed by a 4th-degree permutationally invariant polynomial [Citation97] in variables that are functions of the distances between the ion and the six sites of an MB-pol water molecule. The coefficients of both 2B and 3B permutationally invariant polynomials are optimized using Tikhonov regression [Citation124] (also known as ridge regression) and supervised learning [Citation125] to reproduce the interaction energies calculated at the CCSD(T)-F12b level of theory in the CBS limit [Citation120,Citation126]. It can be demonstrated that 2B and 3B MB-nrg permutationally invariant polynomials effectively recovers quantum-mechanical effects that cannot be described by purely classical expressions (e.g. charge transfer and penetration, and Pauli repulsion) employed in classical polarizable FFs.
4. Many-body effects in halide ion hydration
To provide a systematic analysis of many-body effects in halide ion hydration at the fundamental level, the accuracy of both TTM-nrg and MB-nrg PEFs is assessed through extensive comparisons with reference data for individual terms of the MBE calculated at the CCSD(T)/CBS level as well as with corresponding values calculated with various DFT models that are commonly used in MD simulations of ionic solutions. Also included in these comparisons are results obtained with the AMOEBA polarizable FF [Citation127–Citation130].
4.1 Molecular models and computational details
All reference geometries of the halide-water clusters used in the comparisons shown in the following sections were optimized at the DF-MP2/aug-cc-pVTZ(-PP) level of theory using a strict gradient convergence threshold of 10−6 a.u. Effective core potentials (PP) with 10 and 28 electrons were used for bromide and iodide ions, respectively. For each cluster, all energies were calculated using the stratified approximation many-body (SAMBA) approach [Citation71], using a hierarchy of theory levels for the individual terms in the MBE. Specifically, the MBE terms were calculated using explicitly correlated coupled cluster with single, double and perturbative triple excitations, CCSD(T)-F12b [Citation131,Citation132], a method with successively smaller basis sets for higher-body interactions [Citation133–Citation136]. The CBS limit for the two-body term was achieved by extrapolating the values obtained with the aug-cc-pVTZ(-PP) and aug-cc-pVQZ(-PP) basis sets using a two-point extrapolation [Citation71,Citation137] as in Ref. [Citation120]. The three-body interaction energies were calculated using the aug-cc-pVTZ(-PP) basis sets and corrected for basis set superposition error (BSSE) using the counterpoise method [Citation138]. The four-body and higher-order terms were calculated using the aug-cc-pVTZ(-PP) basis sets without correcting for BSSE. All geometry optimizations and single-point energy calculations were performed using the MOLPRO quantum chemistry software package [Citation139].
For comparison with TTM-nrg and MB-nrg, various DFT models, with and without dispersion contributions [Citation140], are considered in this analysis. GGA (Generalized Gradient Approximation) functionals: BLYP(-D3) [Citation141,Citation142], PBE(-D3) [Citation143], and revPBE(-D3) [Citation144]. Meta-GGA functionals: TPSS(-D3) [Citation145] and SCAN(-D3) [Citation146]. Hybrid GGA functionals: PBE0(-D3) [Citation147], revPBE0(-D3) [Citation144,Citation147], B3LYP(-D3) [Citation148]. Meta-hybrid functionals: M06-2X(-D3) [Citation149]. Range-separated, meta-hybrid functionals: ωB97X [Citation150], ωB97X-D [Citation150], and ωB97M-V [Citation151]. All DFT calculations were carried out with the aug-cc-pVQZ basis using Gaussian 09 [Citation152], except those for revPBE(-D3), SCAN(-D3), revPBE0(-D3), and ωB97M-V that were carried out with Q-Chem [Citation153].
Finally, results obtained with AMOEBA, a generic polarizable FF for biomolecular simulations [Citation127–Citation130], are also included in the comparisons. AMOEBA adopts an induction model with distributed atomic polarizabilities up to the quadrupole moment based on a Thole’s damping scheme and was originally parameterized using both experimental and ab initio data. AMOEBA is under continued development and new versions have recently been proposed [Citation129,Citation130]. All AMOEBA calculations presented were performed with the TINKER package [Citation154], using the AMOEBA 2009 force field [Citation128], which includes the 2003 parameterizations for both water and halide ions [Citation127].
4.2 X–(H2O): Fundamental 2B halide-water interactions
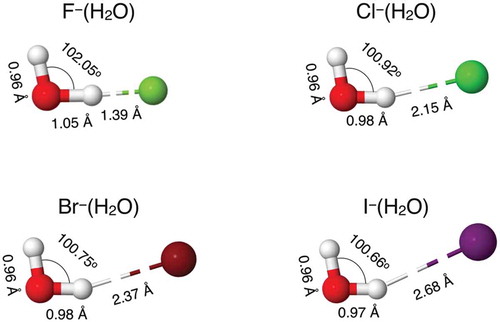
The minimum energy structures predicted by the MB-nrg PEFs for the four X–(H2O) dimers are shown in . Although in all cases the halide ions are found to preferentially form one H-bond with the water molecule, the structural parameters vary appreciably going from F–(H2O) to I–(H2O). In particular, the H-bond increases in length from 1.39 Å to 2.68 Å and becomes less linear, while the HOH angle of the water molecule becomes progressively narrower. These differences can be explained by considering the relatively more pronounced molecular character of HF (or higher basicity of F–) compared to that of other hydrogen halides, which results in more directional and covalent-like interactions between F – and H2O. On the other hand, due to lower and more diffuse charge density, the interactions of iodide with water are primarily electrostatic in nature and dominated by polarization effects.
Figure 1. Minimum energy geometries for X–(H2O) dimers, with X = F, Cl, Br, and I.
The binding energies (Ebind) and harmonic frequencies for the bending (νbend), and H-bonded (νH-bond) and free OH (νfree) stretching modes of the water molecule calculated with both TTM-nrg and MB-nrg PEFs for the four X–(H2O) dimers of are compared in with the corresponding CCSD(T)-F12/aug-cc-pVQZ(-PP) values. In all cases, the values predicted by the MB-nrg PEFs are in quantitative agreement with the reference data. On the other hand, noticeable deviations are found with the TTM-nrg PEFs, which are more pronounced in F–(H2O) and progressively decrease as ion’s size and polarizability increase (and charge density decreases). These differences can be attributed to intrinsic deficiencies associated with the TTM-nrg PEFs which, as all common polarizable FFs, are unable to correctly describe charge transfer and penetration effects through purely classical expressions.
Table 1. Comparison of MB-nrg and TTM-nrg binding energies (in kcal/mol) and harmonic frequencies (in cm−1) calculated for X–(H2O) dimers, with X = F, Cl, Br, and I, with reference data obtained at the CCSD(T)-F12/aug-cc-pVQZ(-PP) level of theory.
It should be noted that fully full-dimensional vibrational spectra calculated for the four X–(H2O) dimers and the corresponding isotopologues, X–(D2O), at the quantum-mechanical level with both TTM-nrg and MB-nrg PEFs have recently been reported [Citation155]. In all cases, the MB-nrg vibrational spectra are in close agreement the the available experimental data, correctly reproducing frequency shifts and tunneling splittings, while significant deviations from the experimental values are found with the TTM-nrg PEFs, which again emphasize the difficulties of purely classical representations in correctly describing halide-water interactions.
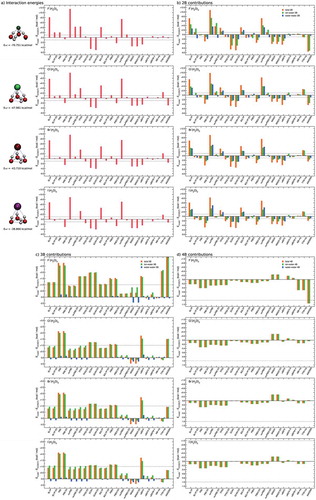
The accuracy of both TTM-nrg and MB-nrg PEFs in reproducing 2B halide-water interactions is further investigated through comparisons with various DFT models as well as with the AMOEBA polarizable FF. Specifically, shows comparisons of deviations relative to CCSD(T)-F12b/CBS interaction energies of the four X–(H2O) dimers calculated for the DF-MP2/aug-cc-pVTZ(-PP) optimized geometries. DFT models, from GGA to range-separated hybrid functionals, are on the left side, while the AMOEBA polarizable FF is on the right side, with TTM-nrg and MB-nrg PEFs in between. As a reference also shown as a dashed line is the ±1 kcal/mol threshold that is generally used to define ‘chemical accuracy’. It should be noted that, in the case of the X–(H2O) dimers, the interaction energies exactly correspond to the 2B term in Eq. 1. As discussed in Ref. [Citation120], a large variability is observed among different DFT models, with GGA and long-range separated hybrid functionals, respectively, providing the poorest and the best agreement with the CCSD(T)-F12b/CBS reference data.
Figure 2. Comparisons between RMSDs associated with various DFT models, TTM-nrg and MB-nrg PEFs, and AMOEBA polarizable FF relative to CCSD(T)-F12b/CBS interaction energies (Eref) calculated for X–(H2O) dimers, with X = F, Cl, Br, and I, in the DF-MP2/aug-cc-pVTZ(-PP) optimized geometries shown on the left.
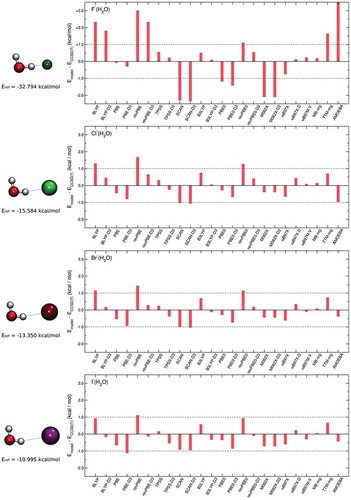
From the analysis of , it is possible to derive general trends that provide guidance in the assessment of different molecular models currently available for halide-water interactions. Overall, the MB-nrg PEF provides the most accurate description of the interaction energies of all X–(H2O) dimers, with the largest deviation being 0.18 kcal/mol for F–(H2O). BLYP and revPBE underestimate the interaction energies of all dimers and the deviations are always larger than 1 kcal/mol, with the exception of I–(H2O) for BLYP. Inclusion of the empirical D3 dispersion correction improves the accuracy of both functionals although the description of F–(H2O) still remains outside the boundaries of ‘chemical accuracy’. Opposite trend is found with the PBE functional that predicts interaction energies within ‘chemical accuracy’ for all dimers and becomes less accurate after inclusion of the empirical D3 dispersion correction, which pushes the deviation for I–(H2O) beyond the 1 kcal/mol threshold. Among the meta-GGA functionals, TPSS predictions are always within 1 kcal/mol while SCAN overestimates the interaction energies of all dimers. In both cases, the inclusion of the D3 dispersion correction appears to have deleterious effects on the ability of these functionals to reproduce 2B interactions. B3LYP, with and without the D3 dispersion correction, is the best performer among all hybrid functionals considered in , while PBE0 is unable to correctly predict the interaction energy of F–(H2O). The D3 dispersion correction has a significant effect on the accuracy of revPBE0, bringing the interaction energy within 1 kcal/mol of the CCSD(T)/CBS reference data for all dimers. Similar to PBE0, the meta-GGA and hybrid M062X functional is not able to correctly reproduce the interaction energy in the F–(H2O) dimer. Interestingly, the D3 dispersion correction has little effect on the performance of M062X. All meta-GGA, hybrid, and long-range separated functionals accurately reproduce the CCSD(T)/CBS reference data, with ωB97M-V consistently providing similar deviations independently of the halide ion.
On the other hand, interaction energies calculated with the TTM-nrg PEF and the AMOEBA polarizable FF are within ‘chemical accuracy’ for all dimers except for F–(H2O), for which both models significantly underestimate the interaction strength. These deviations can be attributed, at least qualitatively, to the inability of purely classical MB induction schemes adopted by polarizable models to correctly account for quantum-mechanical effects such as charge transfer and penetration, and Pauli repulsion, which are emphasized in F–(H2O) by the covalent-like nature of the interactions at short and medium ranges.
4.3 X−(H2O)2: Importance of 3B contributions
Halide dihydrates, X–(H2O)2, are the smallest halide-water systems in which two water molecules can, in principle, be simultaneously H-bonded to each other and to the ion, thus representing ideal systems for characterizing the interplay between ion-water and water-water many-body contributions to the total interaction energies. As shown in , however, the two water molecules are H-bonded to each other in the minimum energy structures of the X–(H2O)2 complexes only in the presence of chloride, bromide, and iodide. This can be explained by considering the different size, charge density, and polarizability of each ion, which, directly affecting the relative strengths of halide-water and water-water H-bonds, progressively stabilize the cyclic configuration, going from F – to I – [Citation120,Citation155].
Figure 3. Comparisons between RMSDs associated with various DFT models, TTM-nrg and MB-nrg PEFs, and AMOEBA polarizable FF relative to CCSD(T)-F12b/CBS interaction energies (Eref) calculated for X–(H2O)2 complexes, with X = F, Cl, Br, and I, in the DF-MP2/aug-cc-pVTZ(-PP) optimized geometries shown on the left.
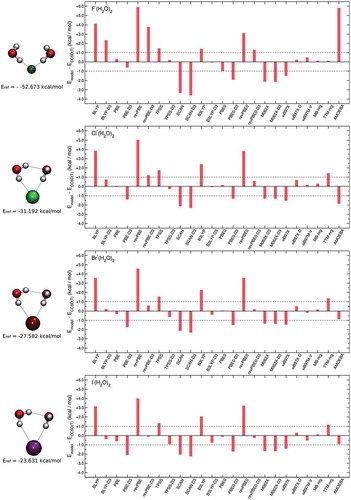
Deviations from CCSD(T)/CBS interaction energies calculated for the minimum energy structures of the X–(H2O)2 complexes also shown in follow similar trends seen for the dimers in Section 4.2. Specifically, the MB-nrg PEFs consistently provide the smallest deviations, while GGA and range-separated, hybrid functionals, including dispersion contributions, respectively, provide the least and most accurate descriptions of the energetics among all DFT models considered in this analysis. From a quantitative point of view, a larger number of DFT models are unable to achieve chemical accuracy than in the case of the X–(H2O) dimers, indicating that halide-water and water-water interactions are effectively described with different levels of accuracy by the same functionals. Finally, both the TTM-nrg PEF and the AMOEBA polarizable FF exhibit larger deviations, on the order of ~1 kcal/mol, for all X–(H2O) complexes except F–(H2O)2, for which TTM-nrg predicts an interaction energy in nearly quantitative agreement with the CCSD(T)/CBS reference data while AMOEBA underestimates the interaction strength by ~6 kcal/mol. The different performances of TTM-nrg and AMOEBA in describing F–(H2O)2 is certainly unexpected since both models represent fluoride-water interactions through similar classical expressions based on many-body induction schemes. These differences thus highlight both difficulties and range of variability associated with polarizable models when applied to molecular interactions that are more quantum-mechanical in nature as F–(H2O)2.
To provide fundamental insights into the performance of different models in representing halide-water systems, the interaction energies of the X–(H2O)2 complexes are further decomposed into individual 2B and 3B contributions associated with ion-water and water-water interactions. The comparisons shown in demonstrate that all models describe 2B water-water interactions relatively more accurately than 2B ion-water interactions, with the latter thus being responsible for the largest fractions of the total deviations from the CCSD(T)-F12b/CBS reference data. Noticeable exceptions are the MB-nrg PEFs and, to some extent, the ωB97M-V functional that represent both 2B ion-water and water-water interactions with similar accuracy. Importantly, some functionals (e.g. BLYP-D3, revPBE-D3, PBE0, revPBE0-D3, and ωB97X-D) appear to benefit from fortuitous error cancellation between 2B ion-water and water-water interactions.
Figure 4. Comparisons between RMSDs associated with various DFT models, TTM-nrg and MB-nrg PEFs, and AMOEBA polarizable FF relative to individual 2B (a) and 3B (b) contributions to interaction energies calculated at the CCSD(T)-F12b/CBS level of theory for X–(H2O)2 complexes, with X = F, Cl, Br, and I, in the DF-MP2/aug-cc-pVTZ(-PP) optimized geometries shown in Figure 3.
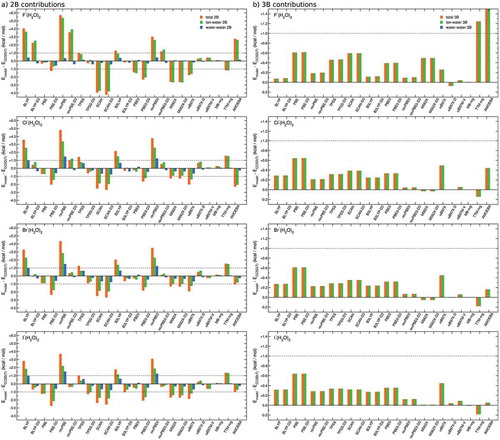
also shows that all models represent 3B ion-water-water interaction within chemical accuracy, with AMOEBA and TTM-nrg effectively providing deviations comparable to those associated with the DFT models, except for F–(H2O)2. In this case, the comparison between TTM-nrg and MB-nrg predictions is particularly insightful. As discussed in Section 3, both models share the same Thole-type induction model with the difference being that this classical term is supplemented in the MB-nrg PEFs by an explicit short-range 3B term expressed as a multidimensional permutationally invariant polynomial (Eq. 7). As shown in , the inclusion of this explicit 3B term allows MB-nrg PEFs to reproduce, nearly quantitatively, the CCSD(T)/CBS reference data, independently of the halide ion. It is also important to note that the 3B ion-water-water deviations associated with both AMOEBA and TTM-nrg systematically decrease from F–(H2O)2, for which they are significantly larger than 1 kcal/mol, to I–(H2O)2. This trend indicates that 3B contributions due to the quantum-mechanical nature of halide-water interactions (e.g. charge transfer and penetration, and Pauli repulsion), which cannot be correctly described by purely classical many-body induction schemes adopted by polarizable FFs, such as AMOEBA and TTM-nrg, play an important role in F–(H2O)2.
4.4 X–(h2o)n=3,4 clusters: Assessment of higher-body contributions
After assessing 2B and 3B effects, in this section, we investigate the role played by 4B and higher-order terms of the MBE in Eq. 1. A comparison between the performance of different models in reproducing the interaction energies of X–(H2O)3 clusters is first analyzed in ). As for the smallest systems, the MB-nrg results are again in close agreement with the CCSD(T)-F12b/CBS reference data, which provides further support for the accuracy of the MB-nrg PEFs. On the other hand, the performance of various DFT models varies significantly depending on the type of functional and nature of the ion. In particular, both positive and negative deviations on the order of several kcal/mol are observed for BLYP, revPBE, SCAN, SCAN-D3, B3LYP, and revPBE0. With the exception of ωB97M-V, whose performance is close to that of the MB-nrg PEFs, all other functionals are associated with deviations larger than 1 kcal/mol at least for one cluster type, with the accuracy of most functionals improving going from F– to I–.
Figure 5. Comparisons between RMSDs associated with various DFT models, TTM-nrg and MB-nrg PEFs, and AMOEBA polarizable FF relative to interaction energies (a), and individual 2B (b), 3B (c) and 4B (d) contributions calculated at the CCSD(T)-F12b/CBS 2B level of theory for X–(H2O)3 clusters, with X = F, Cl, Br, and I, in the DF-MP2/aug-cc-pVTZ(-PP) optimized geometries shown in panel (a).
Defined trends can be established from the analysis of individual contributions to the total interaction energies shown in -d). Several functionals benefit from fortuitous error cancellation between halide-water and water-water contributions. Hybrid functionals overall perform consistently better than GGA functionals. As expected from the analyses of smaller complexes reported in Sections 4.2 and 4.3, the inclusion of the empirical D3 dispersion correction has unpredictable effects on the performance of different functionals in reproducing 2B ion-water and water-water interactions. Deviations on the order of ~1 kcal/mol are in 3B contributions are associated with all functionals, except revPBE0, revPBE0-D3, M06-2X, M06-2X-D3, ωB97X, ωB97X-D, and ωB97M-V. Relatively larger deviations are associated with AMOEBA at all orders.
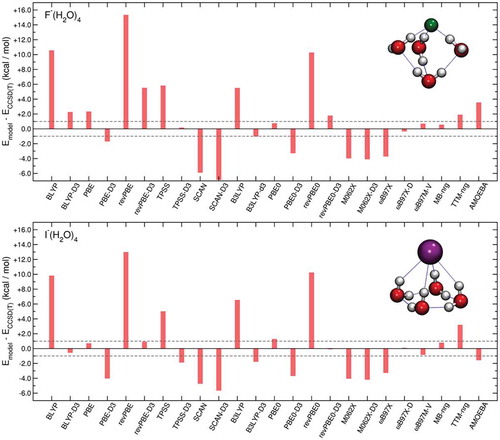
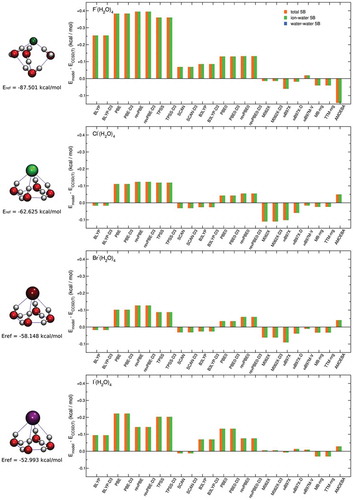
The comparisons shown in -) provides further evidence for the important role played by the 2B and 3B permutationally invariant polynomials in the MB-nrg PEFs which effectively allow the MB-nrg PEFs to achieve a quantitative agreement with the CCSD(T)-F12b/CBS reference data. Importantly, with the exception of AMOEBA results for F–(H2O)3, ) demonstrates that all models considered in this analysis reproduce 4B effects within ‘chemical accuracy’, with both TTM-nrg and MB-nrg PEFs exhibiting the same accuracy as DFT models. This analysis thus indicates that 4B contributions to halide-water interactions are essentially classical-like in nature and can accurately be described through MB induction schemes. , which shows the analysis of 5B contributions to the interaction energies in X–(H2O)4 clusters, provide further evidence for the classical-like nature of higher-body halide-water interactions, demonstrating that both TTM-nrg and MB-nrg PEFs are able to quantitatively reproduce these effects within a classical Thole-type scheme for MB induction.
Figure 6. Comparisons between RMSDs associated with various DFT models, TTM-nrg and MB-nrg PEFs, and AMOEBA polarizable FF relative to 5B contributions to the interaction energies calculated at the CCSD(T)-F12b/CBS 2B level of theory for X–(H2O)4 complexes, with X = F, Cl, Br, and I, in the DF-MP2/aug-cc-pVTZ(-PP) optimized geometries shown on the left.
5. Conclusions and outlook
The last decade has witnessed tremendous progress in the development of accurate molecular models for computer simulations of aqueous systems across different phases. These efforts have been supported by continued growth in computing power as well as by the development of efficient algorithms for correlated electronic structure methods that nowadays enable efficient calculations of interaction energies of small molecular systems, with chemical accuracy. This progress has been accompanied by the development of several approaches to the analytical representation of multidimensional potential energy surfaces [Citation97–Citation99].
Building upon pioneering studies by Stillinger and coworkers [Citation60], and Clementi and coworkers [Citation85,Citation90], and recent advances in the modeling of water from the gas to the condensed phase, in this study we have shown that the many-body expansion of the interaction energy provides a rigorous and efficient approach to the development of analytical potential energy functions for computer simulations of halide-water systems, with chemical accuracy. In particular, through a systematic analysis of the energetics of halide-water clusters, we have demonstrated that the MB-nrg PEFs introduced in Ref. [Citation120] achieve high accuracy by quantitatively reproducing the individual terms of the many-body expansion of the interaction energy by combining explicit short-range representations of 2B and 3B interactions expressed through permutationally invariant polynomials with a physically correct description of many-body electrostatic interactions at all ion-water separations.
Direct comparisons with various DFT models, ranging from GGA functionals to range-separated, meta and hybrid functionals with explicit inclusion of dispersion contributions, demonstrate that most functionals exhibit significant halide-dependent errors in all terms of the many-body expansion of the interaction energy. Among different DFT models, range-separated, meta and hybrid functionals provide the closest agreement with CCSD(T)-F12b reference data. Importantly, the addition of the empirical D3 dispersion correction appears to not necessarily improve the accuracy of the ‘parent’ functionals. On the other hand, our analysis also demonstrates that classical polarizable force fields suffer from intrinsic deficiencies in their functional forms which prevent them from correctly describing short-range contributions to the interaction energies that are quantum mechanical in nature (e.g. charge transfer and penetration, and Pauli repulsion).
Despite representing a step forward toward realistic computer simulations of ionic solutions, it should always be kept in mind that the MB-nrg PEFs are still ‘models’ for ion-water interactions, with their own approximations and limitations. In particular, the MB-nrg PEFs always assume that each halide ion maintains its own chemical identity throughout the simulation, which implies that associated acid-base equilibrium is completely neglected (i.e. for each halide ion X, the corresponding HX acid is always assumed to be a strong acid). While this is a reasonable approximation for Cl–, Br–, and I – in pure water, the situation drastically changes for F – as well as in complex aqueous solutions with pH values lower or higher than 7. Finally, the application of MB-nrg PEFs in computer simulations of complex ionic solutions will require further theoretical and computational developments. In this context, given the unconventional functional form adopted by the MB-nrg PEFs, which is not supported by popular software for computer simulations, synergistic efforts between theoretical/computational chemists/physicists and software engineers will be key to the development of specialized and more efficient software for many-body molecular dynamics simulations which can take full advantage of modern hardware.
Acknowledgments
This work was supported by the National Science Foundation through grants Nos. CHE-1453204, CHE-1305427, and ACI-1642336, and used resources of the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by the National Science Foundation through grant No. ACI-1053575 under allocation TG-CHE110009 on Comet at the San Diego Supercomputer Center and Stampede2 at the Texas Advanced Computing Center. M.R. was supported by a Software Fellowship from the Molecular Sciences Software Institute, which is funded by the National Science Foundation through grant No. ACI-1547580.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Sneen RA. Substitution at a saturated carbon atom. XVII. Organic ion pairs as intermediates in nucleophilic substitution and elimination reactions. Acc Chem Res. 1973;6:554.
- Pregel MJ, Dunn EJ, Nagelkerke R, et al. Alkali–metal lon catalysis and inhibition in nucleophilic displacement reaction of phosphorus–sulfur–and carbon–based esters. Chem Soc Rev. 1995;24:449.
- Nahar S, Tajmirriahi HA. Do metal ions alter the protein secondary structure of a light-harvesting complex of thylakoid membranes? J Inorg Biochem. 1995;58:223.
- Woodson SA. Metal ions and RNA folding: a highly charged topic with a dynamic future. Curr Opin Chem Biol. 2005;9:104.
- Draper DE. RNA folding: thermodynamic and molecular descriptions of the roles of ions. Biophys J. 2008;95:5489.
- Siuzdak G, Ichikawa Y, Caulfield TJ, et al. Evidence of calcium2+-dependent carbohydrate association through ion spray mass spectrometry. J Am Chem Soc. 1993;115:2877.
- Tan ZJ, Chen SJ. Nucleic acid helix stability: effects of salt concentration, cation valence and size, and chain length. Biophys J. 2006;91:518.
- Endo M. Calcium ion as a second messenger with special reference to excitation-contraction coupling. J Pharmacol Sci. 2006;100:519.
- Orlov SN, Hamet P. Intracellular monovalent ions as second messengers. J Membr Biol. 2006;210:161.
- Strater N, Lipscomb WN, Klabunde T, et al. Two-metal ion catalysis in enzymatic acyl- and phosphoryl-transfer reactions. Angew Chem Int Ed. 1996;35:2024.
- Sigel RKO, Pyle AM. Alternative roles for metal ions in enzyme catalysis and the implications for ribozyme chemistry. Chem Rev. 2007;107:97.
- Hanna R, Doudna JA. Metal ions in ribozyme folding and catalysis. Curr Opin Chem Biol. 2000;4:166.
- Stahley MR, Strobel SA. RNA splicing: group I intron crystal structures reveal the basis of splice site selection and metal ion catalysis. Curr Opin Struct Biol. 2006;16:319.
- Harrison RG, Tammet H. Ions in the terrestrial atmosphere and other solar system atmospheres. Space Sci Rev. 2008;137:107.
- Lehtipalo K, Rondo L, Kontkanen J, et al. The effect of acid–base clustering and ions on the growth of atmospheric nano-particles. Nat Commun. 2016;7:11594.
- Winter M, Brodd RJ. What are batteries, fuel cells, and supercapacitors? Chem Rev. 2004;104:4245.
- Jungwirth P, Tobias DJ. Specific ion effects at the air/water interface. Chem Rev. 2006;106:1259.
- Bakker HJ. Structural dynamics of aqueous salt solutions. Chem Rev. 2008;108:1456.
- Marcus Y. Effect of ions on the structure of water: structure making and breaking. Chem Rev. 2009;109:1346.
- Tobias DJ, Stern AC, Baer MD, et al. Simulation and theory of ions at atmospherically relevant aqueous liquid-air interfaces. Annu Rev Phys Chem. 2013;64:339.
- Hofmeister F. Zur lehre von der wirkung der salze. Arch Exp Pathol Pharmacol. 1888;24:247.
- Hofmeister F. Zur lehre von der wirkung der salze. Arch Exp Pathol Pharmacol. 1888;25:1.
- Cox WM, Wolfenden JH. The viscosity of strong electrolytes measured by a differential method. Proc R Soc Lond A Math Phys Sci. 1934;145:475.
- Gurney RW. Ionic processes in solution. McGraw-Hill; 1953.
- Collins KD, Washabaugh MW. The Hofmeister effect and the behaviour of water at interfaces. Q Rev Biophys. 1985;18:323.
- Schmidt DA, Birer O, Funkner S, et al. Rattling in the cage: ions as probes of sub-picosecond water network dynamics. J Am Chem Soc. 2009;131:18512.
- Funkner S, Niehues G, Schmidt DA, et al. Watching the low-frequency motions in aqueous salt solutions: the terahertz vibrational signatures of hydrated Ions. J Am Chem Soc. 2012;134:1030.
- Smith JD, Saykally RJ, Geissler PL. The effects of dissolved halide anions on hydrogen bonding in liquid water. J Am Chem Soc. 2007;129:13847.
- Cappa CD, Smith JD, Wilson KR, et al. Effects of alkali metal halide salts on the hydrogen bond network of liquid water. J Phys Chem B. 2005;109:7046.
- Waluyo I, Nordlund D, Bergmann U, et al. A different view of structure-making and structure-breaking in alkali halide aqueous solutions through x-ray absorption spectroscopy. J Chem Phys. 2014;140:244506.
- O’Brien JT, Prell JS, Bush MF, et al. Sulfate ion patterns water at long distance. J Am Chem Soc. 2010;132:8248.
- Chen Y, Okur HI, Gomopoulos N, et al. Electrolytes induce long-range orientational order and free energy changes in the H-bond network of bulk water. Sci Adv. 2016;2:e1501891.
- Pluharova E, Laage D, Jungwirth P. Size and origins of long-range orientational water correlations in dilute aqueous salt solutions. J Phys Chem Lett. 2017;8:2031.
- Uematsu Y, Bonthuis DJ, Netz RR. Charged surface-active impurities at nanomolar concentration induce Jones–ray effect. J Phys Chem Lett. 2018;9:189.
- Heydweiller A. Density and constitution of watered salt solutions Ann Phys. 1910;33:145.
- Wagner C. The surface tension of dilute solutions of electrolytes. Phys. Z. 1924;25:474.
- Onsager L, Samaras NNT. The surface tension of Debye-Hückel electrolytes. J Chem Phys. 1934;2:528.
- Frumkin A. Phase interface powers and adsorption on the segregative surface air - Solution of anorganic electrolyte. Z. Phys. Chem. 1924;109:34.
- Liu DF, Ma G, Levering LM, et al. Vibrational spectroscopy of aqueous sodium halide solutions and air−liquid interfaces: observation of increased interfacial depth. J Phys Chem B. 2004;108:2252.
- Raymond EA, Richmond GL. Probing the molecular structure and bonding of the surface of aqueous salt solutions. J Phys Chem B. 2004;108:5051.
- Petersen PB, Saykally RJ. Confirmation of enhanced anion concentration at the liquid water surface. Chem Phys Lett. 2004;397:51.
- Perera L, Berkowitz ML. Many‐body effects in molecular dynamics simulations of Na+(H2O)n and Cl−(H2O)n clusters. J Chem Phys. 1991;95:1954.
- Perera L, Berkowitz ML. Structure and dynamics of Cl−(H2O)20 clusters: The effect of the polarizability and the charge of the ion. J Chem Phys. 1992;96:8288.
- Perera L, Berkowitz ML. Stabilization energies of Cl−, Br−, and I− ions in water clusters. J Chem Phys. 1993;99:4222.
- Perera L, Berkowitz ML. Structures of Cl−(H2O)n and F−(H2O)n (n=2,3,...,15) clusters. Molecular dynamics computer simulations. J Chem Phys. 1994;100:3085.
- Dang LX, Smith DE. Molecular dynamics simulations of aqueous ionic clusters using polarizable water. J Chem Phys. 1993;99:6950.
- Stuart SJ, Berne BJ. Effects of polarizability on the hydration of the chloride ion. J Phys Chem. 1996;100:11934.
- Stuart SJ, Berne BJ. Surface curvature effects in the aqueous ionic solvation of the chloride ion. J Phys Chem A. 1999;103:10300.
- Jungwirth P, Tobias DJ. Molecular structure of salt solutions: A new view of the interface with implications for heterogeneous atmospheric chemistry. J Phys Chem B. 2001;105:10468.
- Jungwirth P, Tobias DJ. Ions at the air/water interface. J Phys Chem B. 2002;106:6361.
- Dang LX. Computational study of ion binding to the liquid interface of water. J Phys Chem B. 2002;106:10388.
- Dang LX, Chang TM. Molecular mechanism of ion binding to the liquid/vapor interface of water. J Phys Chem B. 2002;106:235.
- Wick CD, Kuo IFW, Mundy CJ, et al. The effect of polarizability for understanding the molecular structure of aqueous interfaces. J Chem Theory Comput. 2007;3:2002.
- Levin Y, Dos Santos AP, Diehl A. Ions at the air-water interface: an end to a hundred-year-old mystery? Phys Rev Lett. 2009;103:257802.
- Otten DE, Shaffer PR, Geissler PL, et al. Elucidating the mechanism of selective ion adsorption to the liquid water surface. Proc Natl Acad Sci USA. 2012;109:701.
- Baer MD, Mundy CJ. Toward an understanding of the specific ion effect using density functional theory. J Phys Chem Lett. 2011;2:1088.
- Mayer J, Mayer M. Statistical mechanics. New York: John Wiley; 1940.
- Frank HS, Wen WY. Ion-solvent interaction. Structural aspects of ion-solvent interaction in aqueous solutions: a suggested picture of water structure. Discuss Faraday Soc. 1957;24:133.
- Del Bene J, Pople JA. Intermolecular energies of small water polymers. Chem Phys Lett. 1969;4:426.
- Hankins D, Moskowitz JW, Stillinger FH. Water molecule interactions. J Chem Phys. 1970;53:4544.
- Clementi E, Kolos W, Lie GC, et al. Nonadditivity of interaction in water trimers. Int J Quantum Chem. 1980;17:377.
- Yoon BJ, Morokuma K, Davidson ER. Structure of ice Ih. Ab initio two‐ and three‐body water–water potentials and geometry optimization. J Chem Phys. 1985;83:1223.
- Kim KS, Dupuis M, Lie GC, Clementi E. Revisiting small clusters of water molecules. Chem Phys Lett. 1986;131:451.
- Hermansson K. Many-body effects in tetrahedral water clusters. J Chem Phys. 1988;89:2149.
- Xantheas SS, Dunning TH. The structure of the water trimer from ab initio calculations. J Chem Phys. 1993;98:8037.
- Ojamäe L, Hermansson K. Ab initio study of cooperativity in water chains: Binding energies and anharmonic frequencies. J Phys Chem. 1994;98:4271.
- Xantheas SS. Ab initio studies of cyclic water clusters (H2O)n, n=1–6. II. Analysis of many‐body interactions. J Chem Phys. 1994;100:7523.
- Pedulla JM, Vila F, Jordan KD. Binding energy of the ring form of (H2O)6: Comparison of the predictions of conventional and localized‐orbital MP2 calculations. J Chem Phys. 1996;105:11091.
- Hodges MP, Stone AJ, Xantheas SS. Contribution of many-body terms to the energy for small water clusters: a comparison of ab initio calculations and accurate model potentials. J Phys Chem A. 1997;101:9163.
- Cui J, Liu HB, Jordan KD. Theoretical characterization of the (H2O)21 cluster: Application of an n-body decomposition procedure. J Phys Chem B. 2006;110:18872.
- Góra U, Podeszwa R, Cencek W, et al. Interaction energies of large clusters from many-body expansion. J Chem Phys. 2011;135:224102.
- Berendsen HJC, Postma JPM, van Gunsteren WF, et al. Interaction models for water in relation to protein hydration. In: Intermolecular forces. Pullman B, editor. Dordrecht: Reidel; 1981.
- Jorgensen WL, Chandrasekhar J, Madura JD, et al. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926.
- Berendsen HJC, Grigera JR, Straatsma TP. The missing term in effective pair potentials. J Phys Chem. 1987;91:6269.
- Dang LX, Pettitt BM. Simple intramolecular model potentials for water. J Phys Chem. 1987;91:3349.
- Ferguson DM. Parameterization and evaluation of a flexible water model. J Comput Chem. 1995;16:501.
- Mahoney MW, Jorgensen WL. A five-site model for liquid water and the reproduction of the density anomaly by rigid, nonpolarizable potential functions. J Chem Phys. 2000;112:8910.
- Horn HW, Swope WC, Pitera JW, et al. Development of an improved four-site water model for biomolecular simulations: TIP4P-Ew. J Chem Phys. 2004;120:9665.
- Wu YJ, Tepper HL, Voth GA. Flexible simple point-charge water model with improved liquid-state properties. J Chem Phys. 2006;124:024503.
- Paesani F, Zhang W, Case DA, et al. An accurate and simple quantum model for liquid water. J Chem Phys. 2006;125:184507.
- Habershon S, Markland TE, Manolopoulos DE. Competing quantum effects in the dynamics of a flexible water model. J Chem Phys. 2009;131:024501.
- Park K, Lin W, Paesani F. A refined MS-EVB model for proton transport in aqueous environments. J Phys Chem B. 2012;116:343.
- Joung IS, Cheatham TE. Determination of alkali and halide monovalent ion parameters for use in explicitly solvated biomolecular simulations. J Phys Chem B. 2008;112:9020.
- Lybrand TP, Kollman PA. Water–water and water–ion potential functions including terms for many body effects. J Chem Phys. 1985;83:2923.
- Matsuoka O, Clementi E, Yoshimine M. CI study of the water dimer potential surface. J Chem Phys. 1976;64:1351.
- Lie GC, Clementi E. Molecular-dynamics simulation of liquid water with an ab initio flexible water-water interaction potential. Phys Rev A. 1986;33:2679.
- Lyubartsev AP, Laaksonen A. Determination of effective pair potentials from ab initio simulations: application to liquid water. Chem Phys Lett. 2000;325:15.
- Honda K, Kitaura K. A new form for intermolecular potential energy functions. Chem Phys Lett. 1987;140:53.
- Niesar U, Corongiu G, Huang MJ, et al. Preliminary observations on a new water-water potential. Int J Quantum Chem. 1989;36:421.
- Niesar U, Corongiu G, Clementi E, et al. Molecular dynamics simulations of liquid water using the NCC ab initio potential. J Phys Chem. 1990;94:7949.
- Stillinger FH, David CW. Polarization model for water and its ionic dissociation products. J Chem Phys. 1978;69:1473.
- Cisneros GA, Wikfeldt KT, Ojamäe L, et al. Modeling molecular interactions in water: from pairwise to many-body potential energy functions. Chem Rev. 2016;116:7501.
- Zhang CS, Lu C, Jing ZF, et al. AMOEBA polarizable atomic multipole force field for nucleic acids. J Chem Theory Comput. 2018;14:2084.
- Masia M. Ab initio based polarizable force field parametrization. J Chem Phys. 2008;128:184107.
- Guardia E, Skarmoutsos I, Masia M. On ion and molecular polarization of halides in water. J Chem Theory Comput. 2009;5:1449.
- Wick CD. Electrostatic dampening dampens the anion propensity for the air-water interface. J Chem Phys. 2009;131:084715.
- Braams BJ, Bowman JM. Permutationally invariant potential energy surfaces in high dimensionality. Int Rev Phys Chem. 2009;28:577–582.
- Behler J. Perspective: Machine learning potentials for atomistic simulations. J Chem Phys. 2016;145:170901.
- Bartók AP, Payne MC, Kondor R, et al. Gaussian approximation potentials: The accuracy of quantum mechanics, without the electrons. Phys Rev Lett. 2010;104:136403.
- Bukowski R, Szalewicz K, Groenenboom GC, et al. Predictions of the properties of water from first principles. Science. 2007;315:1249.
- Wang YM, Huang XC, Shepler BC, et al. Flexible, ab initio potential, and dipole moment surfaces for water. I. Tests and applications for clusters up to the 22-mer. J Chem Phys. 2011;134:094509.
- Babin V, Medders GR, Paesani F. Toward a universal water model: First principles simulations from the dimer to the liquid phase. J Phys Chem Lett. 2012;3:3765.
- Babin V, Leforestier C, Paesani F. Development of a “first principles” water potential with flexible monomers: Dimer potential energy surface, VRT spectrum, and second virial coefficient. J Chem Theory Comput. 2013;9:5395.
- Babin V, Medders GR, Paesani F. Development of a “first principles” water potential with flexible monomers. II: Trimer potential energy surface, third virial coefficient, and small clusters. J Chem Theory Comput. 2014;10:1599.
- Medders GR, Babin V, Paesani F. Development of a “first-principles” water potential with flexible monomers. III. Liquid phase properties. J Chem Theory Comput. 2014;10:2906.
- Richardson JO, Perez C, Lobsiger S, et al. Concerted hydrogen-bond breaking by quantum tunneling in the water hexamer prism. Science. 2016;351:1310.
- Cole WTS, Farrell JD, Wales DJ, et al. Structure and torsional dynamics of the water octamer from THz laser spectroscopy near 215 μm. Science. 2016;352:1194.
- Brown SE, Götz AW, Cheng X, et al. Monitoring water clusters “melt” through vibrational spectroscopy. J Am Chem Soc. 2017;139:7082.
- Reddy SK, Straight SC, Bajaj P, et al. On the accuracy of the MB-pol many-body potential for water: Interaction energies, vibrational frequencies, and classical thermodynamic and dynamical properties from clusters to liquid water and ice. J Chem Phys. 2016;145:194504.
- Pham CH, Reddy SK, Chen K, et al. Many-body interactions in ice. J Chem Theory Comput. 2017;13:1778.
- Medders GR, Paesani F. Infrared and Raman spectroscopy of liquid water through “first-principles” many-body molecular dynamics. J Chem Theory Comput. 2015;11:1145.
- Straight SC, Paesani F. Exploring electrostatic effects on the hydrogen bond network of liquid water through many-body molecular dynamics. J Phys Chem B. 2016;120:8539.
- Reddy SK, Moberg DR, Straight SC, et al. Temperature-dependent vibrational spectra and structure of liquid water from classical and quantum simulations with the MB-pol potential energy function. J Chem Phys. 2017;147:244504.
- Medders GR, Paesani F. Dissecting the molecular structure of the air/water interface from quantum simulations of the sum-frequency generation spectrum. J Am Chem Soc. 2016;138:3912.
- Moberg DR, Straight SC, Paesani F. Temperature dependence of the air/water interface revealed by polarization sensitive sum-frequency generation spectroscopy. J Phys Chem B. 2018;122:4356.
- Moberg DR, Straight SC, Knight C, et al. Molecular origin of the vibrational structure of ice Ih. J Phys Chem Lett. 2017;8:2579.
- Gaiduk AP, Pham TA, Govoni M, et al. Electron affinity of liquid water. Nat Commun. 2018;9:247.
- Paesani F. Getting the right answers for the right reasons: Toward predictive molecular simulations of water with many-body potential energy functions. Acc. Chem Res. 2016;49:1844.
- Arismendi-Arrieta DJ, Riera M, Bajaj P, et al. i-TTM model for ab initio-based ion–water interaction potentials. 1. Halide–water potential energy functions. J Phys Chem B. 2016;120:1822.
- Bajaj P, Götz AW, Paesani F. Toward chemical accuracy in the description of ion–water interactions through many-body representations. I. Halide–water dimer potential energy surfaces. J Chem Theory Comput. 2016;12:2698.
- Burnham CJ, Anick DJ, Mankoo PK, et al. The vibrational proton potential in bulk liquid water and ice. J Chem Phys. 2008;128:154519.
- Tang KT, Toennies JP. An improved simple model for the van der Waals potential based on universal damping functions for the dispersion coefficients. J Chem Phys. 1984;80:3726.
- Johnson ER, Becke AD. A post-Hartree–Fock model of intermolecular interactions. J Chem Phys. 2005;123:024101.
- Tikhonov A. Solution of Incorrectly Formulated Problems and the Regularization Method. N Soviet Math. 1963;4:1035.
- Abu-Mostafa YS, Magdon-Ismail M, Lin H-T. Learning from data. AML. 2012.
- Riera M, Mardirossian N, Bajaj P, et al. Toward chemical accuracy in the description of ion–water interactions through many-body representations. Alkali-water dimer potential energy surfaces. J Chem Phys. 2017;147:161715.
- Ren PY, Ponder JW. Polarizable atomic multipole water model for molecular mechanics simulation. J Phys Chem B. 2003;107:5933.
- Ponder JW, Wu C, Ren P, et al. Current status of the AMOEBA polarizable force field. J Phys Chem B. 2010;114:2549.
- Wang LP, Head-Gordon T, Ponder JW, et al. Systematic improvement of a classical molecular model of water. J Phys Chem B. 2013;117:9956.
- Laury ML, Wang LP, Pande VS, et al. Revised parameters for the AMOEBA polarizable atomic multipole water model. J Phys Chem B. 2015;119:9423.
- Adler TB, Knizia G, Werner HJ. A simple and efficient CCSD(T)-F12 approximation. J Chem Phys. 2007;127:221106.
- Knizia G, Adler TB, Werner HJ. Simplified CCSD(T)-F12 methods: Theory and benchmarks. J Chem Phys. 2009;130:054104.
- Dunning TH. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J Chem Phys. 1989;90:1007.
- Kendall RA, Dunning TH, Harrison RJ. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J Chem Phys. 1992;96:6796.
- Woon DE, Dunning TH. Gaussian basis sets for use in correlated molecular calculations. III. The atoms aluminum through argon. J Chem Phys. 1993;98:1358.
- Dunning TH, Peterson KA, Wilson AK. Gaussian basis sets for use in correlated molecular calculations. X. The atoms aluminum through argon revisited. J Chem Phys. 2001;114:9244.
- Hill JG, Peterson KA, Knizia G, et al. Extrapolating MP2 and CCSD explicitly correlated correlation energies to the complete basis set limit with first and second row correlation consistent basis sets. J Chem Phys. 2009;131:194105.
- Boys SF, Bernardi F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol Phys. 1970;19:553.
- Werner HJ, Knowles PJ, Knizia G, et al. Molpro: a general-purpose quantum chemistry program package. Wiley Interdiscip Rev Comput Mol Sci. 2012;2:242.
- Grimme S, Antony J, Ehrlich S, et al. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J Chem Phys. 2010;132:154104.
- Becke AD. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys Rev A. 1988;38:3098.
- Lee CT, Yang WT, Parr RG. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B. 1988;37:785.
- Perdew JP, Burke K, Ernzerhof M. Generalized gradient approximation made simple. Phys Rev Lett. 1996;77:3865.
- Zhang YK, Yang WT. Comment on “generalized gradient approximation made simple”. Phys Rev Lett. 1998;80:890.
- Tao JM, Perdew JP, Staroverov VN, et al. Climbing the density functional ladder: nonempirical meta–generalized gradient approximation designed for molecules and solids. Phys Rev Lett. 2003;91:146401.
- Sun JW, Ruzsinszky A, Perdew JP. Strongly constrained and appropriately normed semilocal density functional. Phys Rev Lett. 2015;115:036402.
- Adamo C, Barone V. Toward reliable density functional methods without adjustable parameters: the PBE0 model. J Chem Phys. 1999;110:6158.
- Becke AD. Density-functional thermochemistry. III. The role of exact exchange. J Chem Phys. 1993;98:5648.
- Zhao Y, Truhlar DG. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor Chem Acc. 2008;120:215.
- Chai JD, Head-Gordon M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys Chem Chem Phys. 2008;10:6615.
- Mardirossian N, Head-Gordon M. Mapping the genome of meta-generalized gradient approximation density functionals: the search for B97M-V. J Chem Phys. 2015;142:074111.
- Frisch MJ, Trucks GW, Schlegel HB, et al. Wallingford, CT, USA: Gaussian 09, Gaussian, Inc; 2009.
- Shao YH, Gan ZT, Epifanovsky E, et al. Advances in molecular quantum chemistry contained in the Q-Chem 4 program package. Mol Phys. 2015;113:184–215.
- Rackers JA, Laury ML, Lu C, et al. TINKER 8: a modular software package for molecular design and simulation. 2017.
- Bajaj P, Wang XG, Carrington T, et al. Vibrational spectra of halide-water dimers: Insights on ion hydration from full-dimensional quantum calculations on many-body potential energy surfaces. J Chem Phys. 2018;148:102321.