
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
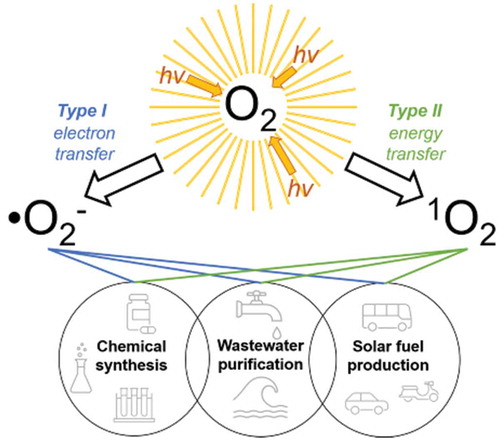
In theory, oxygen (O2) is an ideal chemical reagent because of its high relative abundance and negligible environmental toxicity. In practice however, by the nature of its ground state electronic configuration, many chemical reactions involving O2 are spin forbidden which dramatically decreases its reactivity and thus its utility in applications. More reactive forms of O2 can be achieved by changing its electronic configuration through the use of photochemical and photophysical methods. This review highlights the roll of photon-to-chemical energy conversion in two of these reactive oxygen species (ROS): superoxide (O2−) and singlet oxygen (1O2), which can be accessed through a number of photochemical methods and used in a variety of exciting applications. The theory behind ROS is introduced as produced using light irradiation. Then applications of these methods for chemical transformations are explored.
GRAPHICAL ABSTRACT
1. Introduction
Solar energy has emerged as a front runner in sustainable fuel alternatives as part of an effort to move away from fossil fuel energy consumption. By far, solar energy is the most economical and clean alternative-energy-source, and with advancements in solar technology over the last 40 years, has resulted in light harvesting efficiencies that are up to 10 times greater than even plants [Citation1,Citation2]. Though the surface area that could potentially be dedicated to solar energy harvesting is vast, the dispersion and time-dependency of energy generated from solar technology is still a major problem, and often results in the use of fossil fuels to balance power grids when the sun is not shining. Current challenges in photovoltaics therefore do not lie in capturing sunlight, but in its storage and use.
The field of solar energy storage has been dominated by the use of batteries as a solution to this problem [Citation3,Citation4]. Though advances in battery technology have led to widespread commercial use of solar panel technology coupled with lithium ion battery storage, immense limitations on availability of lithium and long term stability necessitates new technologies. An alternative approach to solar energy storage has led to the idea of solar fuel generation, in which the sun is used to propel a chemical transformation that generates a fuel which can then be used at a later time[Citation5]. In addition, applications of solar-to-chemical energy conversion have been utilized in green chemical synthesis, in which chemical products relevant to industrial processes can be synthesized with light.
One emergent technology is using the unique properties of reactive oxygen species (ROS) generated by light and a co-catalyst to access industrially relevant synthetic targets. Photocatalytic aerobic oxidation reactions are a promising category of solar-to-energy conversion technology because of the inexpensive and environmentally neutral combination of light and oxygen to generate an oxidant. The ROS that have gained the most traction in this field are superoxide (O2−) and singlet oxygen (1O2).
This review will summarize both the fundamentals of O2− and 1O2 generation in aerobic oxidation photocatalysis in addition to providing an overview of the applications of ROS generation in photon-to-chemical energy technologies. The low activity of atmospheric oxygen (3O2) will be described and routes to ROS will be explained via theory and application specifically in aerobic oxidative transformations. We will also comment on and compare the homogeneous and heterogeneous technologies that have emerged through the study of ROS in aerobic oxidation reactions and their practical applications in large scale systems.
2. Fundamentals
2.1. Reactive oxygen species
Atmospheric oxygen exists as a triplet-spin-state in the ground electronic state (X3Σg−)[Citation6]. The degeneracy of the π orbitals of triplet oxygen (3O2) can be visualized in the crude molecular orbital diagram shown in ().
Figure 2.1.1. Molecular orbital diagram of O2 in the ground triplet state, with the two unpaired electrons in the degenerate πp* orbitals outlined in red
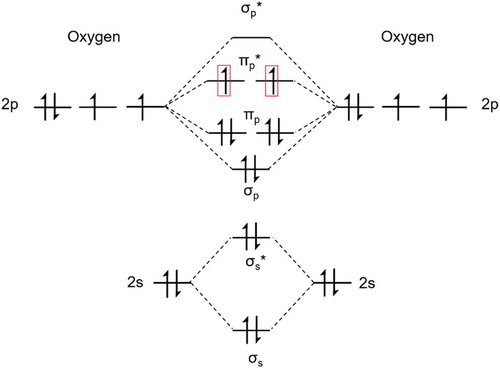
Selection rules dictate that the reaction between molecules in an electronic triplet state and a singlet state is spin forbidden. The spin selection rule states that the electronic spin state of a system must be maintained. Reactions breaking this rule are often kinetically unfavored even if they are thermodynamically probable. In synthetic chemistry applications, the disparity between triplet spin states and organic compounds with singlet spin states results in low reactivity towards oxidation. Applications involving 3O2 reactivity are not covered in the scope of this review.
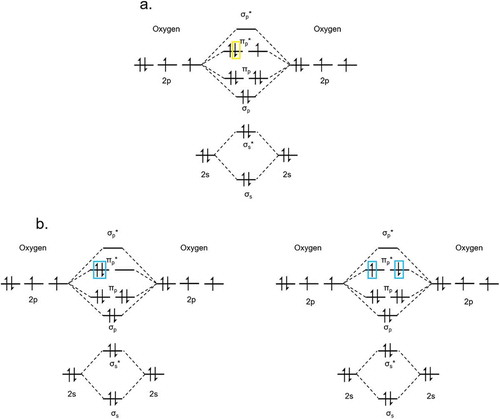
Reactive oxygen species can be generated photochemically from 3O2 through energy transfer (ET) or charge transfer (CT), which occur as intermolecular photophysical processes. O2− is formed as a doublet spin-state with one unpaired electron () via intermolecular CT from a photosensitizer (PS), a compound that upon light absorption is responsible for generating a chemical or electronic change in the acceptor molecule or material. In this case, the PS is first excited to PS*, after which PS* donates its excited electron into one of the two degenerate π* molecular orbitals of 3O2, resulting in the anionic species O2− and cationic PS+. PS+ can be returned to PS in the presence of a sacrificial electron donor or cationic electrode to make a system catalytic in O2− generation.
1O2 is formed via Dexter ET from sufficient orbital overlap with a 1O2 photosensitizer in the triplet excited state (3PS*). Because spin conservation is maintained between 3PS* and 3O2, ET can occur between the two. The mechanism of Dexter energy transfer between 3PS* and 3O2, where 3O2 quenches the triplet excited state of PS, results in the formation of 1O2 as well as 1PS. The singlet electronic configurations of 1O2 are a1Δg (22.5 kcal mol−1) and b1Σg+ (31.5 kcal mol−1) and can be depicted by a crude MO diagram ()[Citation7].
Figure 2.1.2. The molecular orbital diagrams of a) O2− doublet (n = 1) configuration, with one unpaired electron in one of the π* molecular orbitals and b) 1O2 triplet in the two possible electronic configurations
Owing to its utility, 1O2(a1Δg) has a relatively long lifetime both in the gas phase (∼1 h) and in solution (1.5 ± 0.5 s−1 in benzene) as compared to 1O2 (b1Σg+) [Citation8,Citation9]. This can be ascribed to the parity forbidden nature of the transition from a1Δg to b1Σg+, whereas the transition from a1Δg to b1Σg+ can be experimentally observed through phosphorescent emission at 1268.7 nm[Citation10].
2.2. ROS photosensitizers: Type I
Photosensitizers can be categorized based on the type of ROS they generate, and this review will focus exclusively on Type I, O2− producing, and Type II, 1O2 producing, photosensitizers[Citation11]. In the simplest homogeneous systems, organic Type I photosensitizers can be used alone or in conjunction with a single electron redox mediator in order to afford O2−. In these cases, a peroxo-radical (ROO∙) intermediate species is formed when photogenerated organic radical (R∙) reacts with 3O2. This can be achieved in basic aerated alcoholic systems through excitation of the solvent matrix with vacuum-UV photolysis or high-energy ionizing radiation [Citation12,Citation13]. Arguably more conveniently, ketones such as benzophenone and anthraquinone are a commonly used class of redox active aromatic photosensitizers which, when used in conjunction with UV light and primary or secondary alcohols, can produce O2− radicals in basic aqueous conditions [Citation14,Citation15]. Direct electron transfer from an organic redox active substrate to 1O2 can also afford O2−. Substrates including amines, phenols, sulfides, enamines, azines, and azide anions can transfer an electron to photogenerated 1O2, resulting in O2− and the oxidized redox mediator [Citation16–18].
The use of transition metal compounds and materials with the above described redox mediators have also been employed in the generation of O2−, and have provided a more viable route to realizing the use of visible light in O2− generation with longer lifetimes due to a higher resistance to photodegradation. In homogeneous photoredox catalysis, polypyridyl complexes of Ruthenium(II) and Iridium(III) are two of the most commonly used Type I photosensitizers that can engage in single electron transfer processes facilitated by a tertiary amine to generate O2− [Citation19–23]. In these systems, photoinduced metal-to-ligand charge transfer (MLCT) generates the oxidized metal center, *Ru(III) or *Ir(IV) and a reduced ligand. Reductive quenching of the excited state complex via single electron transfer from a tertiary amine affords Ru(I) or Ir(II). Generation of O2− occurs via electron transfer from the reduced complex and returns the photocatalyst to the ground state. Careful tuning of the electronic and structural properties of these organometallic photosensitizers can switch their photoreactivity, allowing them to function as Type I or Type II photosensitizers under different reaction conditions [Citation24,Citation25].
In heterogeneous systems, such as those with commonly studied photoactive materials like TiO2, Bi2WO6, ZnO, or perovskites (PdI2), superoxide generation has been observed occurring at the gas-solid or liquid-solid interface. Photoinduced electron transfer to O2 at metal oxide surfaces such as these can be coupled to an electrode or a sacrificial electron donor electrolyte to effectively scavenge photogenerated holes (h+) to catalytically produce O2−. The literature in this area has been dominated by superoxide generation by TiO2, seeing the use of photoactive powders, films, quantum dots, nanoparticles, and nanoneedles. Modifications of TiO2 surfaces seek to improve rates of reduction and yield of O2− by reducing electron-hole recombination rates and targeting faster charge transport, as well as more favorable light absorption properties of these materials (i.e. red-shifted absorption profiles). Some modifications include doping (both organic and metallic), self-doping, and surface modification via dye adsorption. Perhaps the most relevant application to solar fuel production by modified TiO2 materials is hydrogen peroxide generation via O2− production, which will be discussed below.
2.3. ROS photosensitizers: Type II
Type II photosensitizers possess specific photophysical characteristics that enable them to generate 1O2 via dynamic or static quenching mechanisms. In both cases, a type II photosensitizer has a triplet state (3PS* or T1) that is energetically aligned with that of 1O2 and a long-lived triplet excited state (τT). Though not a requirement for generating 1O2, photosensitizers with a high quantum yield (ΦT) of the excited state triplet are generally favored for synthetic applications.
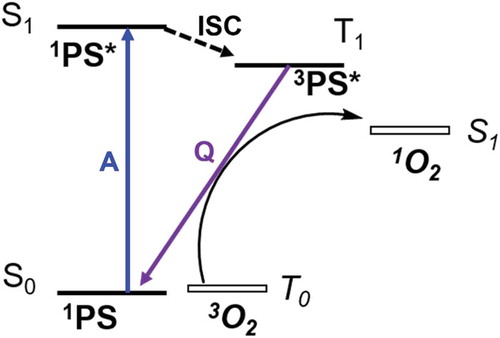
A type II photosensitizer must have an excited state high enough in energy to be able to transfer sufficient energy to 3O2. The energy difference of T0 → S1 transition of oxygen lies in the near IR region at ~1270 nm or 0.98 eV, therefore, the energy difference between S0 and T1 of the PS must be ≥ 0.98 eV in order to generate 1O2. In addition to this, the 3PS* must have a long enough lifetime (τT > 1 µs) to allow for the collision between 3PS* and 3O2 to occur in solution phase systems. Lastly, the rate of intersystem crossing of the PS should be fast, correlating to a high probability of triplet state formation when a photon is absorbed. Energy transfer from PS to 3O2 is commonly visualized with a Jablonski diagram, provided in ().
Figure 2.3.1. Jablonski diagram of the formation of 1O2 from 3O2 quenching the excited state triplet of the photosensitizer (3PS*). Absorption (A) in 1PS forms 1PS*, which can intersystem cross (ISC) to 3PS*. Quenching (Q) of 3PS* by 3O2 then forms 1O2 and reforms 1PS
The efficiency of Type II photosensitizers in generating 1O2 can be described by 1O2 quantum yield (Φ∆), Equation 2.3.1.
where ΦT is triplet quantum yield, [O2] is the dissolved O2 concentration, ken is the rate of energy transfer and kr, knr, and kq are the rates of radiative decay, non-radiative decay, and quenching, all of which dictate energy transfer efficiency. Often, the second term of equation 1 is simplified as Φen, which describes the overall efficiency of energy transfer. The singlet oxygen quantum yields of a variety of photosensitizers, including the ones discussed in this section, are listed below in ().
Table 2.3.1. Compilation of singlet oxygen yields (Φ∆) from various photosensitizers
() includes a variety of detection methods for determining Φ∆. 1O2 can be detected and quantified from its phosphorescent emission at 1270 nm using the appropriate spectroscopic methods (time-resolved infrared luminescence spectroscopy, TRIL), however, indirect methods of detection have also been developed. Compounds that scavenge 1O2 can be used to quantify Φ∆ when coupled to the appropriate spectroscopic methods of product detection. Relevant to the values in (), diphenylisobenzofuran (DPBF) [Citation32,Citation33], dimethylfuran (DMF) [Citation34] and furfuryl alcohol (FFA) [Citation35,Citation36] fall into this indirect detection method category. The change in pressure from 1O2 consumption by dimethylsulfoxide (DMSO) [Citation37] as a 1O2 scavenger has also been recently reported as an inexpensive method of indirectly measuring Φ∆.
In homogeneous photocatalysis, both organic and metal-containing photosensitizers have been developed that can effectively produce 1O2 from 3O2. Phenothiazinium and xanthene dyes are two of the most common types of organic singlet oxygen photosensitizers that absorb a wide range of visible light. Most recognizable of the phenothazinium photosensitizers is methylene blue () with an absorption range of 550–680 nm, giving it a deep blue color in organic solvents.
Scheme 1. Structure of cationic phenothiazinium dyes methylene blue (left), toluidine blue O (center) and new methylene blue (right)
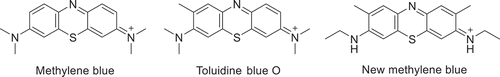
These dyes are characterized by a heteroatomic anthracene core with nitrogen and sulfur atoms located para to one another in the center thiazine ring. Xanthene dyes, i.e. Eosins and Rhodamines (), are another common class of organic singlet oxygen photosensitizers, which can be distinguished by their chalcogen containing xanthene ring located para to an additional phenyl moiety.
Scheme 2. Common xanthene dye derivatives including rose Bengal (top left), fluorescein (top right), eosin blue (bottom left), and rhodamine 6 G (bottom right). Counterions not pictured
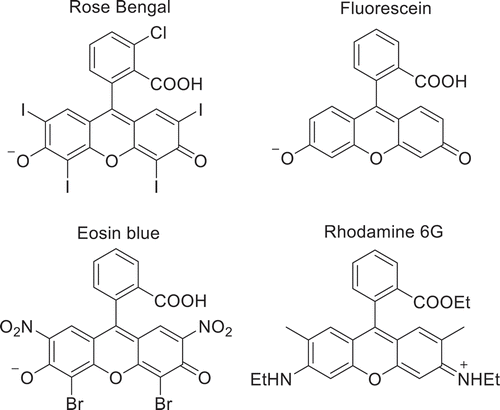
Other organic photosensitizers that have been shown to generate singlet oxygen include aromatic hydrocarbons (naphthalenes, anthracenes and biphenyls) as well as heteroaromatic compounds (quinolines and porphyrins).
As previously described, many organometallic polypyridyl-transition metal complexes also function as Type II photosensitizers. In the absence of a single electron redox mediator, the excited state photosensitizer (1PS*) rapidly intersystem crosses to a metal-to-ligand charge transfer triplet state (3PS*), which can then be quenched by O2 to form 1O2 via energy transfer. Ru(II), Ir(II), and Os(II) metals are most commonly used in these sensitizing complexes, though Cr(III), Pd(II), and Pt(II) complexes have also been reported to produce 1O2 with appropriate photochemical conditions. Porphyrins are also able to sensitize 1O2 in their free-base form or when coordinated to a metal center. Interesting photophysical tunability has been achieved in porphyrin and phthalocyanine derivatives coordinated to a variety of different diamagnetic and paramagnetic metals, including Zn(II), Al(III), Cu(II) and Co(II). In many cases the introduction of a coordinating metal ion to tetraphenylporphine (TPP), a common unmodified porphyrin, improved triplet lifetimes and triplet quantum yield, as well as singlet oxygen quantum yield relative to free-base TPP, improving from 0.62 to 0.65 with Zn coordination ().
Heterogeneous 1O2 generation has focused on developing immobilized singlet oxygen photosensitizers, as many photoexcited semiconductors like TiO2 and ZnO almost exclusively function as Type I photosensitizers. Immobilizing 1O2 photosensitizers has been explored due to the practical applications in technologies like wastewater purification or largescale chemical synthesis in which the photosensitizer must be removed before further processing can occur. Reduction in singlet oxygen yield is observed in these immobilized photosensitizer systems when compared to homogeneous photosensitizers, typically due to a decrease in O2 diffusion through the heterogeneous material. For this application, polymers are an attractive heterogeneous anchoring source, due to the convenience of an organic covalent attachment strategy to immobilize both organic and inorganic photosensitizers. These photosensitizer containing materials have been employed with both organic and organometallic single molecule photosensitizers. The most notable consequence of immobilizing the photosensitizer is a marked decrease in the rate of photosensitizer photobleaching – also likely a consequence of decreased O2 availability in the material – meaning that the photosensitizer containing polymer can be recycled and reused multiple times before significant loss of reactivity is observed. The trend in stability follows that of the aforementioned Type II photosensitizers. Increased photostability is observed in Ru/Ir(bpy)3 and metalloporphyrin photosensitizers immobilized on common polymers like polymethylmethacrylate (PMMT), Amberlite™, and silica when compared to organic photosensitizers like Rose Bengal.
In other heterogeneous solar energy harvesting systems, namely organic photovoltaics, quenching of O2 to form 1O2 results in oxidative degradation of the conjugated material [Citation38–42]. Though developing a fundamental understanding of this degradation has led to improvement of these devices, only the utility of 1O2 in desired oxidative transformations of reactants will be discussed in this review.
Using light to access the reactive and environmentally neutral potential of oxygen marks an exciting research direction in solar energy harvesting technologies. Though these ROS can be accessed directly with high energy radiation, photosensitizers that can convert low energy UV and visible light makes ROS reaction implementation more desirable in an industrial setting. This has been accomplished homogeneously through the development of both organic and metal-containing chromophores with favorable redox and photophysical properties, like high triplet quantum yields and long triplet lifetimes. Though homogeneous aerobic photocatalysis is an important research venue in this area, the transition from homogeneous to heterogeneous ROS generation has made application of this technology feasible at the industry level, and continues to be an important area of research that capitalizes on this unique photophysical phenomenon.
3. Application of ROS generation
Following their generation, ROS can be utilized in a number of different solar energy harvesting technologies that span an impressive breadth of applications. These applications mark the potential of photon-to-chemical energy harvesting that can revolutionize the roll of photochemistry in transforming a variety of industries.
3.1. Fine chemical synthesis
The use of photogenerated ROS in fine chemical synthesis introduces important green synthetic methods to industrial chemical processes. As their name would suggest, ROS possess high kinetic instability, making them incredibly reactive towards organic molecules and have thus been useful in the synthesis of many organic compounds.
When formed, monovalent O2− typically behaves as a radical anion, causing electron and/or oxygen atom transfer to form a desired product. This radical chemical behavior allows for the generation of other reactive oxygen species from O2− like hydroxyl radicals (•OH), hydrogen peroxide (H2O2) and carbon centered peroxide radicals (ROO•), further propagating reactivity and utility. Notably, the oxidation of alcohols to their corresponding aldehydes and carboxylic acids represent an important functional group transformation in organic chemistry that can be achieved using sensitized O2−. The proposed multistep reaction pathway to ketone formation follows a radical mechanism. Initiation occurs between O2− and the primary or secondary alcohol, followed by dismutation and α-carbon deprotonation, as well as formation of a hydroxyl radical and hydrogen peroxide as byproducts.
1O2 has shown arguably greater synthetic utility in fine chemical production owing to its relatively high stereoselectivity and interesting reactivity, which has allowed for large scale implementation with the synthesis of unique substitution patterns. Reactivity towards dienes has been particularly useful in the generation of endoperoxides, which can be used in cascading syntheses to generate useful precursors relevant to the pharmaceutical and materials industries. The general mechanism of formation of endoperoxides is shown in ().
Scheme 3. Formation of endoperoxide from 4 + 2 cycloaddition of 1O2 to a general diene
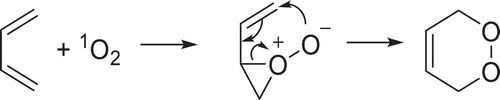
Reaction at one of the alkenes gives a dioxygen epoxide intermediate, followed by an attack of the carbon of the adjacent alkene by the anionic oxygen bound to the peroxide. Opening of the epoxide and migration of the double bond yields two new sigma bonds to the dioxygen moiety. Endoperoxide formation is not restricted to this general diene structure and can be readily formed via reaction of singlet oxygen with cyclic dienes, heterocycles and aromatic rings with applications in cascading biomimetic natural product synthesis. Endoperoxides in furans can be further hydrated to hydroperoxides and further reduced to alcohols, another easily modified functional group. An excellent example of this is in the cascade synthesis of pectenotoxins from their corresponding bi-furan derivatives, which has also been implemented in the total synthesis of Pectenotoxin-2 [Citation43–46]. More recently, this reactivity towards furans has been used in a multi-photocatalyst cascade ()[Citation47].
Scheme 4. Formation of a general bicyclic lactam from furyl-alkyl iodide using methylene blue (MB) and an Ir(III) photocatalyst. Adapted from Angew. Chem. Int. Ed. 2021, 133(8), 4381–4387
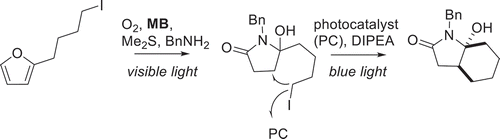
In this case, the singlet oxygen photosensitizer (MB) accomplishes the transformation of furyl-alkyl iodide to a pyrrolone analog with visible light followed by homolytic cleavage of the iodide via electron transfer from the photocatalyst ([(Ir(ppy)2(dtbpy)]PF6). This synthesis strategy afforded the facile production of a variety of complex bioactive alkaloids.
Immobilized organometallic sensitizers have been applied to large scale synthesis of some of the aforementioned natural product targets and have relatively high product yield with facile sensitizer removal and relative ease in photocatalyst recyclability [Citation48–50]. With the improved development of lab scale photocascades, especially with facile, one-pot methods, applications in large scale synthetic efforts will likely increase.
3.2. Wastewater treatment
Though the non-selective and reactive nature of O2− has had less utility in fine chemical synthesis than 1O2, it has proven ideal for detoxification and decontamination of wastewater. Photocatalytic aerobic wastewater treatment has been researched with a large breadth of heterogeneous semiconductor materials including TiO2, CeO2, ZnO, ZrO2, Fe2O3, WO3, V2O5, ZnS, and CdS. Though transition metals have also been explored, their susceptibility to poisoning and deactivation has resulted in minimal representation in wastewater treatment applications, but have seen use as dopants in metal oxide materials to improve visible light absorptivity [Citation51–53]. TiO2 has emerged as a front runner in this research arena, as in other areas of solar energy harvesting, because of its high abundance, low production overhead cost, high stability, and favorable photophysical and electronic properties[Citation54].
As discussed previously in Section 2.2, electrons promoted into the conduction band of TiO2 readily reduce O2 adsorbed to the surface to generate O2− which can then react with a variety bioactive contaminants. O2− formed in situ can also react with H2O to form other oxidizing species like H2O2 and •OH, the oxidation potentials of which are nearly double that of O2− alone. These oxidants expand the scope of substrates that can be decomposed by Type I photosensitizing materials, but only compounds that react with O2− will be discussed.
This reactivity has been particularly effective in the destruction of textile waste dyes. The three most common dyes extruded in wastewater effluent from textile production are Acid Blue 40, Basic Yellow 15 and Direct Blue 87, all of which contain an azo moiety that is reactive towards O2− [Citation55,Citation56]. Near complete degradation of these azo dyes by photocatalytically produced O2− in the presence of UV light and a variety of different TiO2-containing materials has been observed, with decolorization observed in as little as 2 minutes[Citation57]. Similar efficiency has also been seen in O2− mediated degradation of rhodamines, methylene blue, acridine orange, and the reactive dye colors (red, black, and orange), making the realization of zero contaminant wastewater from the textile industry a feasible goal with this technology.
In addition to dyes, TiO2 generated O2− has been used in the treatment of phenols and phenol analogs; organic aromatic compounds containing one or more hydroxyl group deemed one of the most important petrochemical pollutants to remediate from aquatic systems[Citation57]. In addition, the chlorination step involved typical wastewater treatment can generate chlorinated phenol derivatives, which have been shown to be even more persistent in aquatic environments [Citation58,Citation59]. Remediation studies have been carried out with a batch reactor containing heterogeneous TiO2-containing materials (hollow spheres, floating composites, graphite, glass plates, steel fiber, and sand), which when exposed to near-UV radiation (250–350 nm) saw greater than 90% degradation of phenol compounds [Citation60–64]. Typical irradiation times for degradation range from 1–2 hours, with rate dependence on the intensity of the light source and reactant-product equilibrium [Citation65–67]. For photocatalytic aerobic applications of O2 in fine chemical synthesis, where O2− becomes an unwanted biproduct, phenolic compounds are often employed because of their effective and selective scavenging of O2− from solution[Citation68].
1O2 has also shown reactivity towards a variety of organic compounds associated with industrial and municipal wastewater effluent. As previously described, organic photosensitizers have shorter lifetimes in solution compared to immobilized dyes due to 1O2 induced photobleaching. While this presents a challenge to fine chemical synthesis, it has proved surprisingly useful in dye waste remediation. Dyes like Rose Bengal, methylene blue and Eosin Y are Type II photosensitizers, but are also widely used in textile and ink production, where their degradation is necessary to purify the effluent textile wastewater. As a consequence, this has led to interesting research using dyes already present in wastewater to act as remediation photocatalysts. Preliminary experiments based on this observation were accomplished over 30 years ago, but still impact a breadth of waste remediation systems today [Citation17,Citation68–71]. Using self-sensitizing organic waste products present in wastewater has the added benefit of not using an external photosensitizer that requires removal following decontamination.
This innovative strategy has recently been implemented in emergent point-of-use (POU) solar disinfection (SODIS) technologies. Creating simple household and community scale SODIS systems is key to providing clean drinking water to rural populations in the developing world. This mission has been adopted by the Research Center for Nanotechnology-Enabled Water Treatment (NEWT) at Yale University, which has extensively studied the effectiveness of effluent-generated 1O2 in water purification. Type II photosensitizers have been used in SODIS because the oxidative stress caused by 1O2 is effective at killing many bacterial species. Because 1O2 photosensitizers decolorize upon photobleaching, this has been exploited to serve as an indicator of purity in POU application following successful water detoxification. Erythrosine (E) and Riboflavin (RF) are both edible food dyes that are moderately good singlet oxygen photosensitizers (Φ∆[E] = 0.63, Φ∆[RF] = 0.49) and have been used in personal SODIS systems [Citation72–74]. Simulated and natural sunlight irradiation of either of these edible photosensitizers lead to >99% bacteriophage inactivation within 15 minutes. With continued irradiation, photobleaching of the dyes, as indicated by the water changing from colored to colorless, occurred within 30–60 minutes, and thus functioned as a sufficient visual indicator of full bacteriophage inactivation ().
Figure 3.2.1. Time-dependent photobleaching color loss of 5 μM erythrosine with (a) 0 and (b) 30 Nephelometric Turbidity Units (NTU) suspended bentonite clay under natural sunlight (290 W/m2) in 500 mL PET bottles with a complex water composition, with the corresponding . Inactivation of bacteriophage MS2 (c, e) and photobleaching by loss of absorption maximum (d, f) of Erythrosine (red) and Riboflavin (blue). Reprinted figure with permission from Environ. Sci. Technol. 2018, 52, 22, 13,361–13,369. Copyright 2018 American Chemical Society
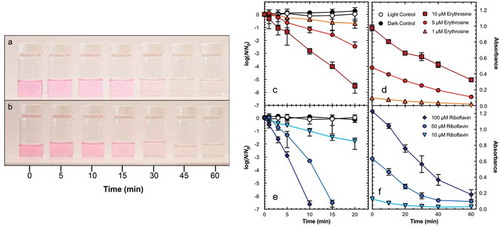
Similarly to O2−, 1O2 can oxidatively react with phenol derivatives in a Diels-Alder like cycloaddition that eventually leads to degradation. Both organic and organometallic Type II photosensitizers immobilized on polymers have been applied to the remediation of phenol and its derivatives. In application, aggregation was a leading cause of poor Φ∆ and rapid photobleaching of the photosensitizers, however, the addition of charged detergents remedied this problem. Complete degradation of phenols and chlorophenols has been observed in the presence of detergent-treated immobilized Rose Bengal, methylene blue and phthalcyanine as well as Al(III), Zn(II), and Ga(III) complexes of phthalcyanines and porphyrins [Citation75,Citation76].
3.3. Hydrogen peroxide generation
Perhaps the most exciting and relevant application of ROS to solar energy harvesting is the photocatalytic aerobic generation of hydrogen peroxide (H2O2) as a solar fuel. The solar fuel field is dominated by research in hydrogen (H2) generation via water splitting. Though H2 is a high energy density and environmentally neutral fuel option that will aid in reducing the emission of greenhouse gases long term, difficulty arises in storage and transport of this gas. In addition, H2 fuel cells have their own implementation challenges, namely that they use platinum, a rare and expensive transition metal as the catalyst, and require a structurally complex proton exchange membrane (PEM) to separate the two half reactions. In contrast, H2O2 when used as a fuel is a 30% aqueous solution, and considerably more compatible with a liquid fuel-based economy. The unique redox behavior of H2O2 allows peroxide fuel cells to function without the need for a membrane and uses an iron catalyst; iron being one of the most abundant and inexpensive metals. With comparable energy output to that of H2 fuel cells (1.09 eV for H2O2 vs. 1.23 eV for H2), H2O2 is arguably a more implementable carbon-free fuel alternative that will reduce dependence on fossil fuels.
Presently, H2O2 is produced via the anthraquinone process, which is energy intensive and uses petrochemically sourced H2 as a starting material. In this process, the energy required to generate H2O2 is greater than the energy generated from a peroxide fuel cell, and is therefore an impractical synthetic approach for generating H2O2 as a fuel. In order to produce H2O2 at the scale necessary to meet current fueling demands, alternative methods for H2O2 generation must be explored. Coupling H2O2 production to solar technology is therefore an extremely important area of research to focus on to realize this goal.
The disproportionation mechanism of H2O2 production from O2− is well understood as a biological process catalyzed in living organisms by the superoxide dismutase enzyme. Under acidic aqueous conditions, O2− generated in solution reacts with available protons to produce HO2 as a highly reactive intermediate, which can then react with itself to form H2O2 and O2 (Equation 3.3.1)[Citation77].
In addition to direct disproportionation, hydroxyl radicals (•OH), a common ROS product of the TiO2 oxygen reduction reaction (ORR), can recombine to generate H2O2. Photoactive metal oxides represent a diverse set of materials that can realize the two-electron photoreduction of O2 to generate H2O2. In addition, using heterogeneous materials in the application of this reaction allows for facile solution purification, as well as ease of solution concentration to meet the 30% aqueous concentration required for a peroxide fuel cell.
Both anitase and rutile unmodified TiO2 surfaces are capable of photocatalytically generating H2O2 with irradiation in the presence of water and O2, however, more efficient H2O2 production has been observed with modified metal oxide materials. As noted in Section 2.2, doping of TiO2 with other compounds leads to increased photostability, improved photophysical properties, and in many cases, improved O2− yield. Though this has been observed with metal ion doped materials, organic doped TiO2 has also been observed to improve H2O2 production in photocatalytic systems. Carbon, nitrogen, sulfur doping in TiO2 have both been extensively studied in waste water purification applications to photocatalytically decompose organic and dye pollutants with H2O2 [Citation78–82].
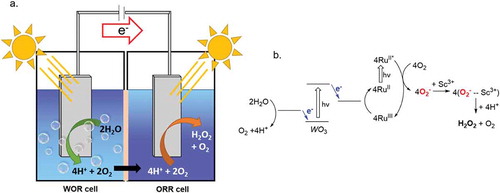
In hybridized heterogeneous systems, photocatalytic H2O2 production via O2 reduction has been coupled to water oxidation in a two-compartment cell system to further improve the sustainability of the process (). In these systems, a water oxidation reaction (WOR) photocatalyst in one cell is responsible for 4-electron water oxidation step. The photoexcited electrons from this step are then used to regenerate the ORR photocatalyst following the formation of O2−.
This process has been accomplished with metal oxide semiconductors as WOR photocatalysts with various modified [Ru(bpy)3]2+ photosensitizers, which generate the O2− necessary to form H2O2[Citation83]. In one system, WO3 and BiVO4 were compared as photoanodes in the WOR half-cell. BiVO4 significantly outperformed WO3, maintaining a current of 0.52 eV vs. SCE from water oxidation after 15 hours of irradiation[Citation84]. In the ORR half-cell, [Ru(bpy)3]2+ could use the photogenerated electrons from the water oxidation reaction in the catalyst regeneration step following O2− production. In this same study, [Ru(Me2phen)3]2+ and [Ru((MeO)2bpy)3]2+ were compared as ORR catalysts to optimize the overall two-compartment cell. Both were observed to have comparable rates of electron transfer, however, UV-Vis experiments showed there was a much larger in situ concentration of Ru3+ in an acidic solution of [Ru((MeO)2bpy)3]2+ (90%) than in [Ru(Me2phen)3]2+ (22%) following continuous irradiation. This resulted in a twofold decrease in the observed photocurrent generated from O2− production. When combined with a Lewis acidic metal to prohibit back electron transfer from O2− to the photocathode, the BiVO4, [Ru((MeO)2bpy)3]2+ and Sc3+ dual compartment cell gave the best performance in photocatalytic production of H2O2 from H2O and O2 ().
Figure 3.3.1. A) Visualization of the two-compartment cell with WOR (left) and ORR (right). b) Catalytic cycle of the photocatalytic production of H2O2 from H2O and O2− using a Ru photocatalyst (RuII, RuII*, and RuIII) and a semiconductor photocatalyst (WO3) facilitated by Sc3+. Adapted from RSC Adv. 2016, 6 (48), 42,041–42,044
The use of Ni and Fe nanoparticles as WOR catalysts have also been explored in similar systems to further progress towards using earth abundant metals as a source of reactivity. Additional work has continued to apply unique materials as the WOR photoanode such as heteropolynuclear complexes, coordination polymers and transition metal decorated metal oxide surfaces coupled to Ru(bpy)3 Type I photocatalysts [Citation85–87].
In an effort to simplify this system even further, metal organic frameworks (MOFs) have been applied to single compartment cells for H2O2 generation. MOFs are heterogeneous crystalline coordination polymers characterized by their uniquely high porosity as well as excellent charge transfer and light harvesting properties. Efficient charge separation in MOFs have made them excellent candidates for photocatalytic applications including CO2 reduction [Citation88], water splitting (ORR [Citation89,Citation90] and WOR [Citation91–93]), and fine chemical synthesis [Citation94,Citation95]. With the more recent shift in focus on solar fuels going beyond photocatalytic H2 production, photoactive MOF research has also expanded to H2O2 generation with unique design strategies for implementation.
As previously stated, the nature of the highly conjugated organic linkers of light active MOFs allows for efficient electron transfer to a variety of substrates. Typically, light active MOFs with these favorable characteristics have a ligand to metal charge transfer (LMCT) excited state, wherein a redox active metal or metal cluster can act as a single electron reductant. Pioneering work by Yamashita and coworkers showed that O2− could be readily produced by photocatalytic electron transfer from MIL-125-NH2, and in the presence of acidic solution, O2− would then disproportionate to H2O2. Following peroxide formation, the photocatalyst can be regenerated with an appropriate anode or sacrificial electron donor, but arguably more interesting is when it can be paired with an industrially relevant organic oxidative transformation, resulting in the photocatalytic production of two important chemical products. This has been accomplished by the Yamashita group and others applying benzyl alcohol oxidation to photocatalytic H2O2 production in MOFs. One particularly interesting implementation of this used MIL-125-NH2 modified with hydrophobic alkyl chains (MIL-125-Rn) to create a biphasic system for separation of the two photocatalytic transformations and isolation of H2O2 ()[Citation96].
Figure 3.3.2. Photocatalytic H2O2 production utilizing the two-phase benzyl alcohol (bottom) and water (top) system. Adapted from Angew. Chem. 2019, 131, 5456–5460
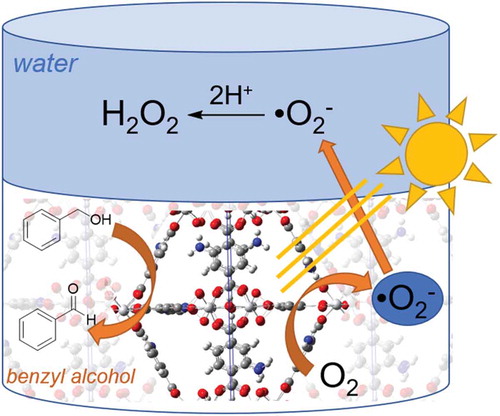
In this two-phase solution of benzyl alcohol and water, H2O2 produced from O2− migrated to the water layer from the organic layer, while the MIL-125-Rn remained in the organic layer and was readily reduced following photoinduced electron transfer by benzyl alcohol to generate benzaldehyde. In addition to ease of separation, this system also helped inhibit the photocatalytic reduction of H2O2 by the photocatalyst, resulting in near fuel level concentrations of aqueous peroxide.
H2O2 production via singlet oxygen generation has been considerably less explored. In practice, using 1O2 to afford H2O2 would be exceptionally useful. The energy transition associated with 1O2 generation means that there is no need for a sacrificial electron donor to regenerate the photocatalyst or protect from back electron transfer, which greatly affect the performance of Type I photocatalysts. In biological systems, the mechanism of H2O2 production via reaction of 1O2 and water catalyzed by antibodies has been termed the antibody catalyzed water oxidation pathway (ACWOP)[Citation97]. Extensive kinetic isotopic labeling experiments and molecular dynamics (MD) simulations have led to a proposed mechanism for ACWOP, in which the antibody uses H2O as an electron source, generating a H2O3 dimer prior to formation of H2O2 and 1O2 or 3O2 [Citation98,Citation99]. Removed from biological systems however, exhaustive literature survey of this area has only afforded H2O2 production from 1O2, water and light in a handful of unique photocatalytic systems.
Tellurium containing compounds and chromophores have exhibited unique reactivity in generating H2O2 from externally sensitized or self-sensitized 1O2. Within a heteroaromatic framework, tellurium exhibits nucleophilic character in a + 2 oxidation state. As such, in the presence of water and 1O2, Te(II) oxidizes to Te(IV), an oxidant species. H2O2 is generated as a biproduct of this oxidation. Most interestingly, this has been observed in self-sensitizing tellurium chromophores, specifically tellurapyrylium and tellurorhodamine dyes, which act as both a Type II photosensitizer and catalyst for the generation of H2O2 [Citation100–102]. The general mechanism of H2O2 production by tellurium-containing dyes is shown in (), where the tellurium active center is highlighted.
Scheme 5. Reaction of 1O2 with a heterocyclic Te center to form H2O2 and the oxidized Te species, a telluroxide.[Citation101]
![Scheme 5. Reaction of 1O2 with a heterocyclic Te center to form H2O2 and the oxidized Te species, a telluroxide.[Citation101]](/cms/asset/4db06ede-02e1-4ba4-b4fa-b49d660ace26/tapx_a_1950049_sch0005_b.gif)
Following generation of self-sensitized 1O2, 1O2 oxidatively adds to the Te center, generating a peroxide intermediate. Hydration of the telluroperoxide forms a hydroxy peroxytellurane, which then forms an equivalent of H2O2 upon formation of the telluroxide[Citation101].
In tellurapyrylium dyes, it has been shown that a second equivalent of H2O2 can be produced upon thermal reduction of Te(IV) to Te(II), generating two equivalents of H2O2 in a single catalytic turnover without the use of a sacrificial electron donor[Citation100]. The interesting and diverse chemistry obtained through the photocatalytic oxidation and reduction of tellurium chromophores is still relatively unexplored, especially in the realm of photocatalytic H2O2 generation and presents a promising avenue for the use of 1O2 in solar fuel production.
4. Conclusions
ROS present a challenging and interesting venue of research to explore photon-to-chemical energy storage that will aid in the move away from a solely petrochemical energy economy. In particular, O2- and 1O2 are two ROS that have seen an increased use in photocatalysis due to their diverse reactivity and ease of photogeneration. Both homogeneous and heterogeneous chromophores have been developed to sensitize O2- or 1O2.
Type I photosensitizers generate O2− via electron transfer from the photosensitizer or photoanode to oxygen in the ground state, 3O2, simultaneously generating a photoexcited hole (h+) that must then be reduced to regenerate the photocatalyst. Both the O2− and h+ generated upon excitation with light have been used in applications of waste water purification and fine chemical synthesis, wherein in situ oxidation and reduction can occur at the sites of charge separation. Heterogeneous applications of O2− in wastewater purification, fine chemical synthesis, and solar fuel production has been dominated by modified TiO2 systems because of ideal photophysical and electronic properties as well as facile photocatalyst removal and recyclability. In some instances, particularly solar fuel production, Type I photocatalysts have been merged with photochemical water oxidation technologies, to make the process of O2− generation even more environmentally sustainable.
Type II photosensitizers undergo Dexter energy transfer to generate 1O2 from 3O2 regenerating the singlet state photosensitizer upon O2 quenching. Homogenous photosensitizers considerably outperform their material counterparts in 1O2 yield which has led to a well-established research field in anchoring photosensitizers to redox active materials. 1O2 has also been used in conjunction with O2− in wastewater purification for its reactivity towards aromatic and heteroaromatic contaminants. In fine chemical synthesis, photogenerated 1O2 has allowed researchers to access synthetically difficult natural products that contain endoperoxide motifs. The full extent of the utility of 1O2 in the production of H2O2 as a solar fuel has yet to be explored, despite its many advantages over O2− in application in this research.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- El Chaar L, Lamont LA, El Zein N. Review of photovoltaic technologies. Renewable Sustainable Energy Rev. 2011;15:2165–25.
- Schlau-Cohen GS. Principles of light harvesting from single photosynthetic complexes. Interface Focus. 2015;5:20140088.
- Li Q, Liu Y, Guo S, et al. Solar Energy Storage in the Rechargeable Batteries. Nano Today. 2017;16:46–60.
- Hou Y, Vidu R, Stroeve P. Solar energy storage methods. Ind Eng Chem Res. 2011;50:8954–8964.
- Gray HB. powering the planet with solar fuel. Nat Chem. 2009;1:7.
- Thatcher Borden W, Hoffmann R, Stuyver T, et al. Dioxygen: what makes this triplet diradical kinetically persistent? J Am Chem Soc. 2017;139:9010–9018.
- Huber KP, Herzberg G. Molecular spectra and molecular structure. US: Springer; 1979. DOI:https://doi.org/10.1007/978-1-4757-0961-2.
- Bregnhøj M, Westberg M, Minaev BF, et al. Singlet oxygen photophysics in liquid solvents: converging on a unified picture. Acc Chem Res. 2017;50:1920–1927.
- Schweitzer C, Schmidt R. Physical mechanisms of generation and deactivation of singlet oxygen. Chem Rev. 2003;103:1685–1757.
- Ogilby PR. Singlet oxygen: there is indeed something new under the sun. Chem Soc Rev. 2010;39:3181–3209.
- Baptista MS, Cadet J, Di Mascio P, et al. Type I and Type II photosensitized oxidation reactions: guidelines and mechanistic pathways. Photochem Photobiol. 2017;93:912–919.
- Varanasi L, Coscarelli E, Khaksari M, et al. Transformations of dissolved organic matter induced by UV photolysis, hydroxyl radicals, chlorine radicals, and sulfate radicals in aqueous-phase UV-based advanced oxidation processes. Water Res. 2018;135:22–30.
- Qiu W, Zheng M, Sun J, et al. Photolysis of enrofloxacin, pefloxacin and sulfaquinoxaline in aqueous solution by UV/H2O2, UV/Fe(II), and UV/H2O2/Fe(II) and the toxicity of the final reaction solutions on zebrafish embryos. Sci Total Environ. 2019;651:1457–1468.
- Núñez Montoya SC, Comini LR, Sarmiento M, et al. Natural anthraquinones probed as Type I and Type II photosensitizers: singlet oxygen and superoxide anion production. J Photochem Photobiol B Biol. 2005;78:77–83.
- Muniyandi K, George B, Parimelazhagan T, et al. Role of photoactive phytocompounds in photodynamic therapy of cancer. Molecules. 2020;25:4102.
- Panzarasa G. Just add luminol to turn the spotlight on radziszewski amidation. ACS Omega. 2018;3:13179–13182.
- Al-Nu’Airat J, Dlugogorski BZ, Gao X, et al. Reaction of phenol with singlet oxygen. Phys Chem Chem Phys. 2019;21:171–183.
- Hayyan M, Hashim MA, Alnashef IM. Superoxide ion: generation and chemical implications. Chem Rev. 2016;116:3029–3085.
- Prier CK, Rankic DA, MacMillan DWC. Visible light photoredox catalysis with transition metal complexes: applications in organic synthesis. Chem Rev. 2013;113:5322–5363.
- Colombo A, Dragonetti C, Guerchais V, et al. A trip in the nonlinear optical properties of iridium complexes. Coord Chem Rev. 2020;414:213–293.
- Zhang Q, Wong KMC. Photophysical, ion-sensing and biological properties of rhodamine-containing transition metal complexes. Coord Chem Rev. 2020;416:213336.
- Liu X, Li G, Xie M, et al. Rational design of Type I photosensitizers based on Ru(Ii) complexes for effective photodynamic therapy under hypoxia. Dalton Trans. 2020;49:11192–11200.
- Bodapati R, Sahoo C, Gudem M, et al. Mononuclear Ru(II) complexes of an arene and asymmetrically substituted 2,2′-bipyridine ligands: photophysics, computation, and NLO properties. Inorg Chem. 2019;58:11470–11479.
- Ruggi A, van Leeuwen FWB, Velders AH. Interaction of dioxygen with the electronic excited state of Ir(III) and Ru(II) complexes: principles and biomedical applications. Coord Chem Rev. 2011;255:2542–2554.
- Alberto ME, Pirillo J, Russo N, et al. Theoretical exploration of Type I/Type II dual photoreactivity of promising Ru(II) dyads for PDT approach. Inorg Chem. 2016;55:11185–11192.
- Le Bechec M, Pigot T, Lacombe S. Chemical quenching of singlet oxygen and other reactive oxygen species in water: a reliable method for the determination of quantum yields in photochemical processes? Chem Photo Chem. 2018;2:622–631.
- Wainwright M, O’Kane C, Rawthore S. Phenothiazinium Photosensitisers XI. Improved toluidine blue photoantimicrobials. J Photochem Photobiol B Biol. 2016;160:68–71.
- Ronzani F, Trivella A, Arzoumanian E, et al. Comparison of the photophysical properties of three phenothiazine derivatives: transient detection and singlet oxygen production. Photochem Photobiol Sci. 2013;12:2160–2169.
- Pereira GFM, Tasso TT. From cuvette to cells: how the central metal ion modulates the properties of phthalocyanines and porphyrazines as photosensitizers. Inorganica Chim Acta. 2021;519:120271.
- Stasheuski AS, Galievsky VA, Stupak AP, et al. Photophysical properties and singlet oxygen generation efficiencies of water‐soluble fullerene nanoparticles. Photochem Photobiol. 2014;90:997–1003.
- Sikorska E, Khmelinskii I, Komasa A, et al. Spectroscopy and photophysics of flavin related compounds: riboflavin and Iso-(6,7)-riboflavin. Chem Phys. 2005;314:239–247.
- Krieg M. Determination of singlet oxygen quantum yields with 1,3-diphenylisobenzofuran in model membrane systems. J Biochem Biophys Methods. 1993;27:143–149.
- Entradas T, Waldron S, Volk M. The detection sensitivity of commonly used singlet oxygen probes in aqueous environments. J Photochem Photobiol B Biol. 2020;204:111787.
- Oelckers S, Hanke T, Röder B. Quenching of singlet oxygen in dimethylformamide. J Photochem Photobiol A Chem. 2000;132:29–32.
- Haag WR, Hoigné J, Gassman E, et al. Singlet oxygen in surface waters - Part I: furfuryl alcohol as a trapping agent. Chemosphere. 1984;13:631–640.
- Allen JM, Gossett CJ, Allen SK. Photochemical formation of singlet molecular oxygen (1O2) in illuminated aqueous solutions of p-aminobenzoic acid (PABA). J Photochem Photobiol B Biol. 1996;32:33–37.
- Lutkus LV, Rickenbach SS, Mccormick TM. Singlet oxygen quantum yields determined by oxygen consumption. Journal of Photochemistry and Photobiology A: Chemistry, Volume 378, Pages 131-135.
- Weu A, Kumar R, Butscher JF, et al. Energy Transfer to a stable donor suppresses degradation in organic solar cells. Adv Funct Mater. 2020;30:1907432.
- Mateker WR, McGehee MD. Progress in understanding degradation mechanisms and improving stability in organic photovoltaics. Adv Mater. 2017;29:1603940.
- Perthué A, Fraga Domínguez I, Verstappen P, et al. An efficient and simple tool for assessing singlet oxygen involvement in the photo-oxidation of conjugated materials. Sol Energy Mater Sol Cells. 2018;176:336–339.
- Perthué A, Gorisse T, Santos Silva H, et al. Influence of traces of oxidized polymer on the performances of bulk heterojunction solar cells. Mater Chem Front. 2019;3:1632–1641.
- Speller EM, Clarke AJ, Aristidou N, et al. Toward improved environmental stability of polymer: fullerene and polymer: nonfullereneorganic solar cells: a common energetic origin of light- A Nd oxygen-induced degradation. ACS Energy Lett. 2019;4:846–852.
- Kouridaki A, Montagnon T, Kalaitzakis D, et al. Using singlet oxygen to synthesize the CDE-ring system of the pectenotoxins. Org Biomol Chem. 2013;11:537–541.
- Halim R, Brimble MA, Merten J. Synthesis of the ABC fragment of the pectenotoxins. Org Lett. 2005;7:2659–2662.
- Kouridaki A, Sofiadis M, Montagnon T, et al. Pectenotoxin’s ABCDE ring system: a complex target to test the potential of singlet oxygen super cascades as tools for synthesis. European J Org Chem. 2015;2015:7240–7243.
- Fujiwara K, Suzuki Y, Koseki N, et al. Total Synthesis of Pectenotoxin-2. Angew Chem. 2014;126:799–803.
- Kalaitzakis D, Bosveli A, Sfakianaki K, et al. Multi‐photocatalyst cascades: merging singlet oxygen photooxygenations with photoredox catalysis for the synthesis of alkaloid frameworks. Angew Chem. 2021;133:4381–4387.
- Ghogare AA, Greer A. Using singlet oxygen to synthesize natural products and drugs. Chem Rev. 2016;116:9994–10034.
- Esser P, Pohlmann B, Scharfet H-D. Die Photochemische Synthese von Feinchemikalien Mit Sonnenlicht. Angew Chem. 1994;106:2093–2108.
- Funken KH, Ortner J. Technologies for the solar photochemical and photocatalytic manufacture of specialities and commodities: a review. Zeitschrift fur Phys Chemie. 1999;213:99–105.
- Minero C, Pelizzetti E, Sega M, et al. The role of humic substances in the photocatalytic degradation of water contaminants. J Dispers Sci Technol. 1999;20:643–661.
- Zang Y, Farnood R. Photocatalytic activity of AgBr/TiO2 in water under simulated sunlight irradiation. Appl Catal B Environ. 2008;79:334–340.
- Subba KV, Lavédrine B, Boule P. Influence of metallic species on TiO2 for the photocatalytic degradation of dyes and dye intermediates. J Photochem Photobiol A Chem. 2003;154:189–193.
- Wold A. Photocatalytic properties of TiO2. Chem Mater. 1993;5:280–283.
- Ganesh R, Boardman GD, Michelsen D. Fate of azo dyes in sludges. Water Res. 1994;28:1367–1376.
- Tang WZ, Huren A. UV/TiO2 photocatalytic oxidation of commercial dyes in aqueous solutions. Chemosphere. 1995;31:4157–4170.
- Al-Mamun MR, Kader S, Islam MS, et al. Photocatalytic activity improvement and application of UV-TiO2 photocatalysis in textile wastewater treatment: a review. J Environ Chem Eng. 2019;7:103248.
- Pedroza AM, Mosqueda R, Alonso-Vante N, et al. Sequential treatment via trametes versicolor and UV/TiO2/ruxsey to reduce contaminants in waste water resulting from the bleaching process during paper production. Chemosphere. 2007;67:793–801.
- Dillert R, Bahnemann D, Hidaka H. Light-induced degradation of perfluorocarboxylic acids in the presence of titanium dioxide. Chemosphere. 2007;67:785–792.
- Hosseini SN, Borghei SM, Vossoughi M, et al. Immobilization of TiO2 on perlite granules for photocatalytic degradation of phenol. Appl Catal B Environ. 2007;74:53–62.
- Li X, Wang D, Cheng G, et al. Preparation of polyaniline-modified TiO2 nanoparticles and their photocatalytic activity under visible light illumination. Appl Catal B Environ. 2008;81:267–273.
- Sboui M, Cortés-Reyes M, Swaminathan M, et al. Eco-friendly hybrid paper–AgBr–TiO2 for efficient photocatalytic aerobic mineralization of ethanol. Chemosphere. 2021;269:128703.
- Song J, Wang X, Bu Y, et al. Photocatalytic enhancement of floating photocatalyst: layer-by-layer hybrid carbonized chitosan and Fe-N- codoped TiO 2 on fly ash cenospheres. Appl Surf Sci. 2017;391:236–250.
- Gyulavári T, Kovács K, Kovács Z, et al. Preparation and characterization of noble metal modified titanium dioxide hollow spheres – new insights concerning the light trapping efficiency. Appl Surf Sci. 2020;534:147327.
- Kashif N, Ouyang F. Parameters effect on heterogeneous photocatalysed degradation of phenol in aqueous dispersion of TiO2. J Environ Sci. 2009;21:527–533.
- Deepracha S, Ayral A, Ogawa M. Acceleration of the photocatalytic degradation of organics by in-situ removal of the products of degradation. Appl Catal B Environ. 2021;284:119705.
- Ijaz M, Zafar M. Titanium dioxide nanostructures as efficient photocatalyst: progress, challenges and perspective. international journal of energy research. John Wiley and Sons Ltd. International Journal of Energy Research. March 10 2021. 3569–3589. DOI:https://doi.org/10.1002/er.6079.
- Ma J, Zhou H, Yan S, et al. Kinetics studies and mechanistic considerations on the reactions of superoxide radical ions with dissolved organic matter. Water Res. 2019;149:56–64.
- Tratnyek PG, Elovitz MS, Colverson P. Photoeffects of textile dye wastewaters: sensitization of singlet oxygen formation, oxidation of phenols and toxicity to bacteria. Environ Toxicol Chem. 1994;13:27–33.
- Tai C, Zhang S, Wang J, et al. Solar-induced generation of singlet oxygen and hydroxyl radical in sewage wastewaters. Environ Chem Lett. 2017;15:515–523.
- Nassar SJM, Sirbu D, Harriman A. Photocatalysed decolouration of indigo in solution: via in situ generation of an organic hydroperoxide. Photochem Photobiol Sci. 2019;18:2875–2883.
- Ahmad I, Fasihullah Q, Vaid FHM. A study of simultaneous photolysis and photoaddition reactions of riboflavin in aqueous solution. J Photochem Photobiol B Biol. 2004;75:13–20.
- Redmond RW, Gamlin JN. A compilation of singlet oxygen yields from biologically relevant molecules. Photochem Photobiol. 1999;70:391–475.
- Ryberg EC, Chu C, Kim JH. Edible dye-enhanced solar disinfection with safety indication. Environ Sci Technol. 2018;52:13361–13369.
- Nowakowska M, Kępczyński M. Polymeric photosensitizers 2. photosensitized oxidation of phenol in aqueous solution. J Photochem Photobiol A Chem. 1998;116:251–256.
- Gerdes R, Bartels O, Schneider G, et al. Photooxidations of phenol, cyclopentadiene and citronellol with photosensitizers ionically bound at a polymeric ion exchanger. Polym Adv Technol. 2001;12:152–160.
- Bielski BHJ, Allen AO. Mechanism of the disproportionation of superoxide radicals. J Phys Chem. 1977;81:1048–1050.
- Wawrzyniak B, Morawski AW. Solar-light-induced photocatalytic decomposition of two azo dyes on new TiO2 photocatalyst containing nitrogen. Appl Catal B Environ. 2006;62:150–158.
- Nishijima K, Ohtani B, Yan X, et al. Incident light dependence for photocatalytic degradation of acetaldehyde and acetic acid on S-doped and N-doped TiO2 photocatalysts. Chem Phys. 2007;339:64–72.
- Mondal K, Gupta A. Recent advances in carbon–semiconductor nanocomposites for water remediation. In energy, environment, and sustainability. Singapore: Springer Nature; 2018. 45–74. Doi:https://doi.org/10.1007/978-981-10-7551-3_4
- Noman MT, Ashraf MA, Ali A. Synthesis and applications of nano-TiO 2 : a review. Environmental science and pollution research. Springer Verlag;February 8 2019. 3262–3291. DOI:https://doi.org/10.1007/s11356-018-3884-z.
- He HY, Tian CY. Rapid photo- and photo-fenton-like catalytic removals of malachite green in aqueous solution on undoped and doped TiO2 nanotubes. Desalin Water Treat. 2016;57:14622–14631.
- Shibata S, Suenobu T, Fukuzumi S. Direct synthesis of hydrogen peroxide from hydrogen and oxygen by using a water-soluble iridium complex and flavin mononucleotide. Angew Chemie Int Ed. 2013;52:12327–12331.
- Isaka Y, Yamada Y, Suenobu T, et al. Production of hydrogen peroxide by combination of semiconductor-photocatalysed oxidation of water and photocatalytic two-electron reduction of dioxygen. RSC Adv. 2016;6:42041–42044.
- Zhuang H, Yang L, Xu J, et al. Robust photocatalytic H2O2 production by octahedral Cd3(C3N3S3)2 coordination polymer under visible light. Sci Rep. 2015;5:16947.
- Edwards JK, Freakley SJ, Lewis RJ, et al. Advances in the direct synthesis of hydrogen peroxide from hydrogen and oxygen. Catal Today. 2015;248:3–9.
- Kobayashi H, Teranishi M, Negishi R, et al. Reaction mechanism of the multiple-electron oxygen reduction reaction on the surfaces of gold and platinum nanoparticles loaded on titanium(IV) oxide. J Phys Chem Lett. 2016;7:5002–5007.
- Chen Y, Wang D, Deng X, et al. Metal-organic frameworks (MOFs) for photocatalytic CO2 reduction. Catal Sci Technol. 2017;7:4893–4904.
- Wang X, Zhou J, Fu H, et al. MOF derived catalysts for electrochemical oxygen reduction. J Mater Chem A. 2014;2:14064–14070.
- Delaporte N, Rivard E, Natarajan SK, et al. Synthesis and performance of MOF-based non-noble metal catalysts for the oxygen reduction reaction in proton-exchange membrane fuel cells: a review. Nanomaterials. 2020;10:1947.
- Kirner JT, Finke RG. Water-oxidation photoanodes using organic light-harvesting materials: a review. J Mater Chem A. 2017;5:19560–19592.
- Blakemore JD, Crabtree RH, Brudvig GW. Molecular catalysts for water oxidation. Chem Rev. 2015;115:12974–13005.
- Shao Q, Yang J, Huang X. The Design of water oxidation electrocatalysts from nanoscale metal–organic frameworks. Chem A Eur J. 2018;24:15143–15155.
- Subudhi S, Rath D, Parida KM, et al. Approach towards the photocatalytic organic transformations over functionalised metal organic frameworks: a review. Catal Sci Technol. 2018;8:679–696.
- Zhang T, Metal-Organic LW. Frameworks for artificial photosynthesis and photocatalysis. Chem Soc Rev. 2014;43:5982–5993.
- Isaka Y, Kawase Y, Kuwahara Y, et al. Two‐phase system utilizing hydrophobic metal–organic frameworks (MOFs) for photocatalytic synthesis of hydrogen peroxide. Angew Chem. 2019;131:5456–5460.
- Wentworth PJ, Jones LH, Wentworth AD, et al. Antibody catalysis of the oxidation of water. Science (80). 2001;293:1806–1811.
- Datta D, Vaidehi N, Xu X, et al. Mechanism for antibody catalysis of the oxidation of water by singlet dioxygen. Proc Natl Acad Sci U S A. 2002;99:2636–2641.
- Nieva J, Wentworth P. The antibody-catalyzed water oxidation pathway - a new chemical arm to immune defense? Trends Biochem Sci. 2004;29:274–278.
- Detty MR, Gibson SL. Tellurapyrylium dyes as catalysts for the conversion of singlet oxygen and water to hydrogen peroxide. J Am Chem Soc. 1990;112:4086–4088.
- Lutkus LV, Rettig ID, Davies KS, et al. Photocatalytic aerobic thiol oxidation with a self-sensitized tellurorhodamine chromophore. Organometallics. 2017;36:2588–2596.
- You Y, Ahsan K, Detty MR. Mechanistic studies of the tellurium (II)/ Tellurium (IV) redox cycle in thiol peroxidase-like reactions of diorganotellurides in methanol. J Am Chem Soc. 2003;125:4918–4927.