
ABSTRACT
Biological ion channels precisely control the flow of ions across membranes in response to a range of physical and chemical stimuli. With their ability of transporting ions in a highly selective manner and of integrating regulatory cues, they are a source of inspiration for the construction of solid-state nanopores as sensors or switches for practical applications. Here, we summarize recent advancements in understanding the mechanisms of ion permeation and gating in channel proteins with a focus on the elementary steps of ion transport through the pore and on non-canonical modes of intramolecular communication between peripheral sensory domains and the central channel pore.
Graphical Abstract
Introduction
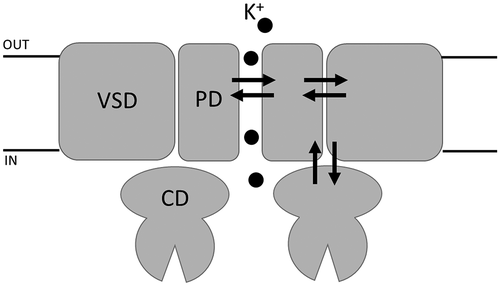
Ion channels are membrane proteins that catalyze the selective and regulated diffusion of ions across cellular membranes. Their activity is mutually integrated into physiological processes of cells in either direction: ion channels control cell signaling pathways and are tightly controlled by them. For this, their main biophysical properties, selectivity and gating, are extremely versatile and tightly and precisely controlled by endogenous and exogenous stimuli. Ion channels contain sensory domains that perceive physical or chemical signals and convert them into electrical activity /ion fluxes that initiate signaling cascades. Hence, ion channels act as miniaturized switches that convert stimuli into electrical activity changes. With these functional properties, ion channels serve as a blueprint for the development of artificial biosensors in which the current flow through pores in solid state materials is modulated by analytes of interest. While solid state nanopores have the advantage of being robust compared to biological systems composed of delicate membranes and proteins, their selectivity for ions, as well as their modulation by ligands, lag behind that of channel proteins, which exhibit extraordinary degrees of selectivity for the transported ions that has been tailored by evolution. One way of improving their performance is to take inspiration from structure/function correlates of ion channels. Ion channel studies are presently advancing very fast due to the constant supply of high-resolution protein structures provided by single particle cryogenic electron microscopy (cryoEM). In combination with computational methods and functional data on single channel activity, investigators are constantly uncovering basic physical and chemical principles underlying their function. The next goal is to derive from a wealth of experimental data, elementary principles in ion channel function and to implement them into artificial systems. Here, we present a small collection of studies addressing basic principles of structure/function correlates in potassium (K+) channels recently presented at the Symposium on “Frontiers in ion channels and nanopores: theory, experiments, and simulation’, held in Rome, 2–5 February 2021. The first two contributions are focusing on the elementary steps of ions flowing through the pore with insights on permeation and gating in K+ channels. Potassium (K+) channels contain one common building element, the so-called pore module (PD) [Citation1]. This highly conserved domain forms, in the functional tetramer, the water-filled central pore for ion transport. This domain includes two transmembrane helices connected via an external loop (turret) and the pore loop (P-loop). The selectivity filter (SF) arises from four re-entrant P-loops, each containing a highly conserved signature sequence, TVGYG, that allows the passage of naked ions without their hydration shell, and where K+ ions are selected against Na+ or other ions. The pore module further controls gating, i.e. opening and closing of the ion permeation pathway, by processing the inputs of the sensory domains which are connected to it by linkers [Citation1]. Each pore forming alpha subunit of Voltage gated K+ channels (KVs) contains six membrane spanning segments (S1–S6) that co-assemble as tetramers to form a fully functional channel. Segments S1 to S4 form the voltage sensor domain (VSD), while segments S5 and S6 form the pore domain (PD). Four PDs congregate radially to form the conductive pathway that is surrounded by four VSDs. Each VSD is directly connected to the PD by a S4-S5 linker, in addition to other mechanisms discusses here as well. Some KVs can be further regulated by ligands. In the case of Hyperpolarization-activated cyclic nucleotide-gated (HCN) channels, the soluble cyclic nucleotide binding domain (CNBD) is attached to the S6 of the PD via a C-linker.
Two other contributions of this review will provide molecular insights in gating mechanisms, i.e. how peripheral sensing domains for voltage and ligand transmit their information to the pore gate.
1-Permeation mechanisms in KcsA channel (Luigi Catacuzzeno, Fabio Franciolini)
The bacterial KcsA has been among the most studied K+ selective channels, and the first to have its structure solved by X-ray crystallography [Citation2]. Soon after the resolution of the crystal structure, the group of Christopher Miller performed electrophysiology experiments on KcsA channels [Citation3] and found sublinear current–voltage (IV) relationships, asymmetry between inward and outward currents, and saturating conductance vs K+ concentration (gC) relationships ()), properties also typical of many other K+ channels.
Figure 1. Mechanisms of K permeation through K+ channels. (A) Experimental data from [Citation3] showing the electrophysiological properties of KcsA channels. Left: Current–voltage relationships obtained at varying K+ concentrations. Right: conductance–concentration relationship assessed at two different voltages. (B) Three-dimensional structure of the KcsA channel, as revealed by X-ray crystallography [Citation2]. C) High resolution electron density map of the KcsA selectivity filter, as obtained (under permission) by [Citation4], showing four putative K+ binding sites inside the selectivity filter, and three additional ones immediately outside. (D) Drawing showing the two main K+ permeation mechanisms proposed for the K+ channel, known as soft and hard knock-on. (E) Two different rate models of K+ permeation are shown, together with their prediction in terms of the current-voltage and conductance–concentration relationships. While the classical knock-on type mechanism predicts hyperlinear current–voltage relationships and non-saturating gCs, an association–dissociation model predicts permeation features quite similar to those experimentally observed for K+ channels (cf panel A). (F) Mathematical relationships connecting the kinetic rate constants of a rate model to the energetic profile (U) and the diffusion constant (D). The expression used for estimating the rate constants not involving the binding of K+ ions was derived from the mean first passage time theory, while the expression for the K+ association binding constants was derived from the Smolukowsky equation
![Figure 1. Mechanisms of K permeation through K+ channels. (A) Experimental data from [Citation3] showing the electrophysiological properties of KcsA channels. Left: Current–voltage relationships obtained at varying K+ concentrations. Right: conductance–concentration relationship assessed at two different voltages. (B) Three-dimensional structure of the KcsA channel, as revealed by X-ray crystallography [Citation2]. C) High resolution electron density map of the KcsA selectivity filter, as obtained (under permission) by [Citation4], showing four putative K+ binding sites inside the selectivity filter, and three additional ones immediately outside. (D) Drawing showing the two main K+ permeation mechanisms proposed for the K+ channel, known as soft and hard knock-on. (E) Two different rate models of K+ permeation are shown, together with their prediction in terms of the current-voltage and conductance–concentration relationships. While the classical knock-on type mechanism predicts hyperlinear current–voltage relationships and non-saturating gCs, an association–dissociation model predicts permeation features quite similar to those experimentally observed for K+ channels (cf panel A). (F) Mathematical relationships connecting the kinetic rate constants of a rate model to the energetic profile (U) and the diffusion constant (D). The expression used for estimating the rate constants not involving the binding of K+ ions was derived from the mean first passage time theory, while the expression for the K+ association binding constants was derived from the Smolukowsky equation](/cms/asset/92173efa-952c-48d1-8aac-e8faf8341a76/tapx_a_1978317_f0001_oc.jpg)
Atomic structure of the KcsA channel
The KcsA channel pore is formed by the juxtaposition of four identical subunits, each composed of two TM segments ()). The SF amino acids (TVGYG) have their carbonyl (and hydroxyl in the case of threonine) oxygens pointing towards the center of the pore.
A successive high resolution electron density map of the KcsA SF clearly identified four high electron density positions (called sites 1 to 4, going from extracellular to intracellular) and few others immediately outside [Citation4] (), left). Unfortunately, it was not possible to establish whether the observed electron densities inside the SF originated from K+ ions or from water, due to their very similar X ray diffraction pattern. However, crystallized channels obtained in presence of Tl ions, that permeate K+ channels and have a much higher number of electrons, suggested a mean ion occupancy at each internal site of the SF close to 0.5, indicating that K+ is found in half of the sites at each given time [Citation5]. This observation was in accordance with several previous studies on unidirectional K+ fluxes [Citation6–8] and streaming potentials [Citation9].
Knock-on mechanism of K+ permeation
Based on these observations, the SF was proposed to mostly reside in either of two possible configurations, containing two K+ ions either at positions 1:3 or 2:4, and water molecules in between. In this view, permeation would occur with a K+ ion or a water approaching from either side of the membrane and pushing forward the single file of K+ and waters, in a mechanism usually referred to as soft knock-on (), top) [Citation4].
In recent years, many experimental and computational data have been pointing to a different mechanism of K+ permeation, in that the SF would never be occupied by water, at least at relatively high bath K+ concentrations. First, crystallographic data in presence of Tl+ ions have been reinterpreted giving an absolute occupancy close to one for all four SF sites [Citation10]. Second, diffracting the KcsA crystal with X-ray wavelengths close to the K+ ions absorption range, a condition used to distinguish K+ from water, confirmed that the SF is fully occupied by K+ ions [Citation11]. Third, solid state nuclear magnetic resonance clearly indicated that a portion of the SF never sees water molecules, meaning that the permeation process does not involve water [Citation12]. Finally, in long molecular dynamics (MD) simulations the passage of water through the SF was never seen [Citation10]. These data suggested the hard knock-on permeation mechanism, meaning absence of intervening water between K+ ions inside the SF, and incoming K+ ions promoting the advancement of the ions inside the SF by a direct (ion-to-ion) interaction (), bottom).
Although these permeation mechanisms, based on structural considerations and MD results, are mechanistically interesting, they only consist of verbal descriptions of the permeation process. That is, they can neither be checked against the experimental results (IV and gC relationships) nor be always validated by all atom MD simulations as ionic current prediction can currently be done only on a limited number of experimental situations due to the high computational expense. Thus, a strategy to test the suggested permeation mechanisms against available experimental data is urgently needed.
Using rate models to describe K+ permeation
More quantitative and predictive mechanisms of permeation could be obtained using rate models where the channel pore is assumed to dwell in few stable configurations, and the process of permeation pictured as ions hopping from a stable configuration to the next, with a probability given by the rate constant characterizing that process [Citation13]. Although rate models can only give an approximate picture of the permeation process, they are able to connect to experimental results through mathematical expressions of ion fluxes, and thus they could be used to evaluate the soundness of a permeation process. In ), two possible rate models of permeation for the KcsA channel are shown, together with their predictions. The first one describes the classic knock-on type permeation mechanism, as proposed by [Citation7], whereby the incoming K+ ion transfers its kinetic energy to the single file inside the pore, making it advance and promote permeation. It can be easily shown that when described mathematically with the use of a rate model, this mechanism generates currents with hyperlinear IV relationships, and it does not predict saturation of the current at high K+ concentrations (), top). Notably, neither of these predictions are consistent with the experimental results of permeation through KcsA channels.
By contrast, an association/dissociation (A/D) mechanism would be able to produce the right output, as clearly described by [Citation14], and illustrated in ), bottom. In these types of model, current is produced by the binding of a K+ ion to the SF on one side of the membrane and the unbinding of another K from the other side. In this case, the exit of the K+ ion is temporally separated from the binding of a K+ ion on the other side. In contrast to classical knock-on mechanisms, A/D models do predict sub-linear IV relationships and gC curves with a Michaelis-Menten saturation (), bottom), suggesting that they might be more appropriate to describe permeation through KcsA and other K+ selective channels.
A common and direct approach to verify the validity of a rate permeation model is to fit experimental current data obtained at different voltages and ion concentrations and see if the fitting values obtained can reproduce the IVs and gC relationships (see MODEL BASED FITS in section 2 below). This approach suffers, however, from two limitations. First, it does not give any hint of the underlying model. Second, the fitting values obtained might only represent the combination of rate constants that give a relative minimum in the fitting function, thus not representing the true rate constants. As an alternative procedure, we present below a multi-scale approach that uses molecular dynamics (MD) data for choosing the model and numerical values of the rate constant and experimental data to test the predictive value of the selected model.
Using multi-scale approach for a quantitative description of permeation mechanisms
A strategy to identify a realistic mechanism of K+ permeation through KcsA channel could make use of a multi-scale approach, whereby the information obtained from MD is first used to define a plausible rate model of permeation, in terms of both the number, types and connections between the stable configurations considered, and the quantitative values of the rate constants connecting them. Once this step is completed, the rate model can be easily verified for its predictive capability of the permeation properties experimentally available. This approach appears to be applicable on a broad scale, as the rate constants of the resulting model can easily be derived from MD data. Rate constants not involving the binding of a K+ ion can be estimated as the inverse of the mean first passage time (MFPT), that is, the time needed to go from the initial state (i) to the final state (f) of the process [Citation15, b; Citation16]. Conversely, with rate constants involving K+ association a possibility is to make the reasonable assumption that they are diffusion limited and assess their values with the equation shown in ), right, coming from the Smolukowsky equation. If a more rigorous estimate of the association rate constant is needed, Brownian/MD approaches can be considered [Citation17].
The mathematical relationships reported in ) show that the rate constant depends on the energy profile encountered during the process and the diffusion constant characterizing the transition of the system from one configuration to the other. Notably, both these parameters can easily be obtained from all atom MD [Citation18,Citation19]. Thus, a multi-scale approach using permeation rate models inspired by MD results could represent a useful strategy to verify the correct predictions of a postulated permeation mechanism in KcsA as well as other ion channels.
Finally, it needs to be mentioned that a similar multi-scale approach could also be used by combining MD with coarse-grained models of K+ permeation other than rate models, such as Brownian models where structural features and electrostatics can be more explicitly be incorporated [Citation20,Citation21].
2-The physical chemistry of gating in a K+ channel pore (Oliver Rauh, Indra Schroeder, Gerhard Thiel)
Structure/function correlates in K+ channels are from the functional side achieved by single channel recordings, which allow to measure the stochastic switching between open and closed states of different gates with high (tens of µs) temporal precision [Citation22]. Complementary structural insights are provided by a wealth of high-resolution K+ channel structures and computational simulations [Citation23,Citation24]. A causal relationship between single channel data and information on channel structures and simulations should eventually lead to a picture in which the dwell times of experimentally determined individual open or closed states can be associated with a stochastic conformational change or chemical reaction in a channel protein.
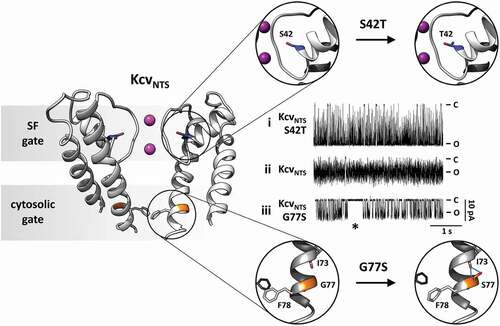
To identify the molecular ABC of gating in the pore module of K+ channels we use small and simple K+ channel proteins from viruses [Citation25–27]. From an experimental and analytical point of view, they are well suited for this endeavor because with about 80 amino acids (AA) per monomer (KcsA has 160 AA), they are about one order of magnitude shorter than mammalian channels (). This greatly reduces the level of system complexity and hence supports its understanding. In spite of their small size, they exhibit all the structural elements of the pore module of complex K+ channels as well as their basic functional features including selectivity and gating. This allows cross referencing to available K+ channel structures and extrapolation to structure/function correlates in complex K+ channels.
Figure 2. Location and function of two distinct gates in model K+ channel pore Cartoon representation of KcvNTS in side-view orientation with two opposing monomers (left). The selectivity filter (SF) gate causes fast flicker gating in KcvNTS with the effect that the full open state is not fully resolved (trace ii). The mutation S42T in the pore helix behind the selectivity filter (S42T: upper part of right panel) slows down fast gating with the result that the full open state of the channel is achieved (trace i). The presence (trace iii) or absence (trace ii) of a long-lived closed state (*) as result of the cytosolic gate depends on presence or absence of S77 in inner transmembrane helix (G77S: lower part of right panel). The cytosolic gate is operated by the formation/breaking of an intrahelical H-bond and the consequent re-orientation of F78. Single channel currents were recorded with symmetrical 100 mM KCl (10 mM HEPES/KOH pH 7.0) in DPhPC bilayers. C and O highlight the closed and open levels, respectively
Systematic Dwell Time Analysis
Small viral K+ channels are ideal for a systematic single channel analysis. They can be synthesized in vitro and inserted into nanodiscs from which they are easily reconstituted into planar lipid bilayers for single-channel recordings [Citation27]. The systematic and highly reproducible recordings of the reference channel KcvNTS show that it has one open and four distinct closed states [Citation26,Citation27]. Two closed states are in the range of milliseconds and can be extracted from conventional dwell-time analysis (). The remaining two closed states have time constants, which are below the resolution limit of typical electrical recording systems [Citation26]. These fast-closed events can be obtained analytically by the so-called extended beta distribution analysis, which was developed for this purpose [Citation22].
The question of the structural features responsible for the four distinct closed states can be addressed by comparing the single-channel gating in structurally similar but not identical channels. An interesting candidate is KcvS, which differs only in 11 AAs from the reference channel KcvNTS [Citation27]. Even these AA exchanges are not dramatic in nature but mostly conservative (8 AAs) or semiconservative (3 AAs) substitutions. Single-channel recordings of KcvS show one additional very long closed state over KcvNTS; the other states are rather similar to that of the latter [Citation27]. Extensive mutational analysis and single-channel recordings have identified the structural nature of this long-lived closed state. It is based on AA S77 at the end of the inner transmembrane helix. MD simulations of homology structures of both channels confirm that the critical Ser forms an intrahelical hydrogen-bond with the amide oxygen of AA I73 upstream in the helix, which in turn induces a slight change in the peptide torsion angles of the helix turn between I73 and S77 [Citation27] (). The resulting kink in the inner transmembrane helix moves the side chain of F78 in the same subunit away from the central axis. Complementary MD simulations suggest that kink formation occurs only in two opposing subunits [Citation27]. This symmetry breaking makes space for Phe side chains of the neighboring subunits to fully move into the pathway and block ion flow. Consequently, the channel is in the long-lived closed state as long as S77 is forming the intrahelical hydrogen bond and maintains a structural asymmetry. The equivalent Phe side chains in KcvNTS are unable to form this closed state, because they mutually obstruct each other for entering this closed state forming configuration.
The important message from this is that channel gating can be based on very simple chemical interactions such as bifurcated hydrogen bond formation in an alpha helix. Worth noting is that intrahelical hydrogen-bonds and kinks in transmembrane domains are not rare in proteins in general including ion channels. Hence, gating does not necessarily require large conformational changes. Furthermore, the presence/absence of a gate in a channel can be caused by rather mild AA substitutions. In the case of the intrahelical hydrogen-bond formation, only Ser and Thr are suitable AAs.
Pore gating
A feature of all Kcv type channels is a flicker gating of the inward currents. The lifetime of the open events decreases in a voltage dependent manner with hyperpolarization to such an extent that they are no longer fully resolvable in conventional single-channel recordings [Citation22,Citation25–27] (). This artificial filter function of the recording system results in an apparent decrease in the unitary conductance at negative voltages. The technical artifact can be overcome by analytical approaches in which the noise of the open channel is used in combination with fitting of realistic gating models to recover the true channel conductance as well as the rate constants for opening and closing [Citation22,Citation26]. Detailed analysis of this voltage-dependent gating has shown that it is dominated by a voltage-dependent closing of the channel [Citation25,Citation26]. Many experimental evidences relate this gate to the selectivity filter (SF) of the channel. Single-channel data show that voltage dependency of the transition into the short-lived closed state is sensitive to the concentration of the transported K+ ions [Citation25,Citation26]. This fosters the view that fast closed events result from depletion or change in the occupation probability of K+ in SF binding sites. The interpretation of fast closing events in the context of SF gating is also supported by mutational analysis and structural considerations. It occurred that the mutation S42T in the pore helix of KcvNTS caused a suppression of the aforementioned voltage-dependent fast closed events [Citation25,Citation26] (). The I/V relation of this mutant is linear and shows no longer the negative slope conductance generated by the fast gate. The relationship between this mutation and pore gating is not only supported by its location in the region of the SF () but by the fact that the critical amino acid is in the same position as E71 in KcsA [Citation28,Citation29]. Mutations of this AA are able to modulate SF gating by corrupting a salt bridge between the pore helix and the SF [Citation29]. In the case of KcvNTS, S42 is not involved in salt bridge formation but is part of a hydrogen-bond network between pore helix and SF. Hence, while the chemical nature of the pore helix/ SF contacts in KcsA is different from those in Kcv, the functional properties are conserved.
Model-based fits
The challenge in understanding structure/function correlates in K+ channels is to associate distinct open and closed states from experimental studies under different experimental conditions with the structural dynamics of the respective channel proteins. A complementary method to coarse grained and prolonged MD simulations is provided by what we call model-based fitting [Citation25]. The idea behind this method is that the shapes of single channel I/V relations under different experimental conditions must reflect the molecular process of permeation. Permeation models for the flow of K+ ions through the SF of a channel can be derived from a combination of high-resolution structural data and MD simulations. Based on a wealth of these data from the literature, there are suitable models for the hopping of K+ ions between the SF binding sites S0 to S4 [Citation25]. These permeation models can be translated into kinetic models and used to fit experimental data [Citation25]. This approach is suitable for testing model predictions from structural and computational studies and can also be used for relating experimental findings to structural entities in a channel protein.
We used this approach for relating the fast-closing events of SF gating in Kcv channels, to the ion permeation in K+ channels [Citation25]. This system is ideal for employing this method since the literature provides structures with atomistic resolution on the SF of K+ channels and because fast filter gating is linked to ion permeation [Citation25]. Because the I/V relation of a channel is the consequence of permeation the best model should provide the best fit to multiple I/V relations obtained under many different experimental conditions. The latter includes a large range of voltages and different symmetrical and asymmetrical K+ concentrations on both sides of the membrane. For fitting, three reasonable permeation models with four or five states and with or without water between the permeating K+ ions, were used. Best results were yielded with a five-state model, which includes not only the four binding sites S1-S4 but also S0 [Citation25]. The latter site was identified as sensor for the external K+ concentration, a parameter, which affects the voltage dependency of fast filter gating. By fitting different models, a “soft knock on” permeation, e.g. with water between the ions, was superior to the alternative “direct knock on’ model [Citation25]; in the latter, ions are permeating in single file without water between ions. At this point, it is important to say that this approach allows for a testing of model predictions; the data do not rule out a direct knock on permeation of ions through the SF. But for explaining the experimental data, the structural model needs to be modified from the standard model used in this approach.
3-Noncanonical gating mechanism in voltage-gated K+ channels (Joao Luis Carvalho-de-Souza, Carlos A Z Bassetto Jr and Francisco Bezanilla)
Voltage-gated ion channels combine ion selectivity and voltage dependence to give cells electrical excitability. These types of ion channels enable cells to fire action potentials, which are transient depolarizations of the membrane negative resting potential. Voltage-gated potassium channels (VGKC) open at membrane potentials more positive than the resting potential thus repolarizing the cell membrane during action potentials. Even in non-excitable cells, VGKC keep the membrane potential negative. VGKC are normally homotetrameric protein complexes formed by alpha subunits (KVs) associated with regulatory subunits. Both the ion selectivity and the voltage dependence mechanisms reside in the alpha subunits and for this reason the great majority of the structure-function studies and the development of modulators are directed to KVs.
In the voltage sensor domain (VSD, S1-S4) of KVs, the S4 segment contains charged amino acids (sensing charges), usually arginine. These residues are exposed to a focused electric field from the membrane electric potential [Citation30,Citation31]. At the resting potential (negative inside), the charged residues reside in the intracellular region. Upon depolarization, these charges drag S4 vertically towards the extracellular side with rotation, translation and change in tilt [Citation32,Citation33].
The movement of the sensing charges can be recorded as gating currents, which are about two orders of magnitude smaller than the ionic currents. The channel activation gate is in the pore domain (S5-S6) and it is formed by crossing of the S6 segments from all four subunits of the tetrameric functional channel. The voltage-induced movements of S4 segment are transferred to S5, by the S4-S5 linker, which ultimately change the position of the S6 segments. The simplified VSD-to-PD coupling mechanism described above comprises the great majority of the information flowing from the VSD to the PD during membrane potential changes that gates the channel and it is the canonical electromechanical coupling mechanism [Citation34, Citation35, Citation41].
Recently, structural biology studies combined with molecular dynamics and functional assays have revealed other coupling mechanisms in VGKC that are called noncanonical coupling [Citation36–39]. These noncanonical mechanisms do not depend on the S4-S5 linker-based canonical coupling mentioned above. Rather, it is based on the extensive and non-covalent contact interface between the segments S1 or S4, and S5. In a model of VGKC, S4 and S5 segments are shown in very close proximity and their side chains are within the van der Waals distance from each other [Citation40]. Hence, suggesting a physical mechanism whereby S4 movements are directly transferred to S5.
To date, two architectural designs in VGKC have been determined based on the relative position of VSDs and PDs. In some KV families (KV1-4 and KV7), the VSD and the PD are swapped between adjacent subunits, yielding a interface formed by S4 and S5 from different subunits [Citation40–42]. In other KV families (KV 10–12), there is no domain-swapping and the S4 and S5 interface is formed by structures from the same subunit [Citation43,Citation44].
We have observed anomalous VSD behavior in a mutant of Shaker K+ channel (Shaker), a channel from fruit fly homolog to the human KV1 family, therefore a domain-swapped channel Shaker [Citation45] has been extensively studied as archetype of VGKC. In Shaker, the W434F mutation in the P-loop is extensively used to abrogate ionic conduction by accelerating slow inactivation, thus allowing the recording of gating currents [Citation46,Citation47]. This mutation, although being in the PD, was shown to interfere with the VSD dynamics when associated with the mutation L361R in the VSD. The resting state of the VSD containing L361R was stabilized by W434F mutation, and the result was a crossing of the charge vs voltage (Q-V) with the conductance vs voltage (G-V) curves. Interestingly, these two mutated residues locate more than 14 angstroms away from each other. At that time, accidentally, we have uncovered what we thought to be a possible alternative (noncanonical) communication between the PD and the VSD, other than the canonical coupling mechanism [Citation45].
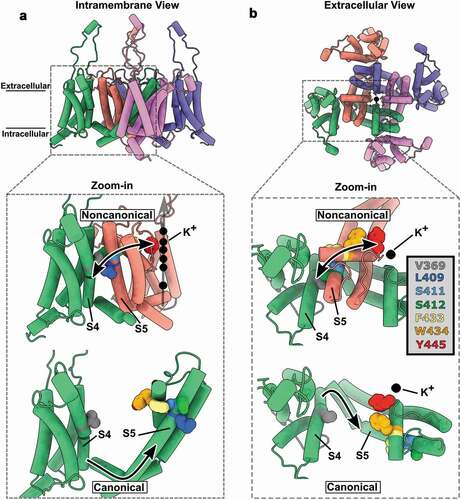
We decided to investigate this phenomenon in depth, and the fact that Shaker features domain-swapping architecture helped us to come out with a way to study the noncanonical coupling mechanism. We designed and produced tandem dimers of Shaker and added mutations in the VSD of one protomer and in the PD of the other protomer [Citation37]. This strategy allowed us to capture the interaction between the VSD and the PD through the noncanonical mechanism when the channel was expressed as dimers of dimers, as opposed to tetramers of monomers. We then demonstrated that the VSD endows voltage sensitivity to the PD, especially to the selectivity filter in the form of inactivation, through the interface between a VSD from one subunit (protomer) and the PD from another subunit (another protomer). By looking at the structural models recognized to apply to Shaker [Citation40], we proposed a group of residues that the Van der Waals surfaces of their side chains are in close proximity as the molecular basis for this novel noncanonical coupling mechanism. The string of amino acids displays a pathway parallel to the membrane plane. The implied residues are leucine (L409), serine (S411 and S412), phenylalanine (F433) and tryptophan (W434). They are in S5 and P-loop region and form a direct connection between the VSD and the tyrosine (Y445) located at the selectivity filter ().
Figure 3. Molecular basis for the noncanonical coupling mechanism in Shaker K+ channel. (A) Intramembrane view of the active state of the voltage sensor domain (VSD) and open state of the pore domain (PD) in a model of Kv1.2, a human Shaker K+ channel homolog (PDB:3LUT). (B) Extracellular view of A showing swapped domains in the channel complex. The zoomed views show the color-coded residues V369, L409, S411, S412, F433, W434 and Y445 that are implicated in the noncanonical mechanism that couples VSD and PD from distinct subunits (also color coded). The upper zoomed views show the physical (and functionally tested) connection between a VSD from one subunit and a PD from a different subunit which is the molecular basis of the noncanonical coupling mechanism. The bottom zoomed views show the same residues but, as they are in the same subunit, they are uncoupled to the S4 segment
Recently, we uncovered another case where the noncanonical mechanism is playing a role. A single mutation in the S4 VSD (L366H) that relieves the inactivation induced by W434F [Citation36]. We then designed a set of experiments intended to probe whether the hypothesized chain is the molecular basis for the noncanonical coupling mechanism in Shaker. We mutated each of the chained residues that we had proposed into alanine in order to change its fundamentals: the amino acid residues volume. Whenever we interrupted the chain (shown in ), we produced conspicuous changes on how the VSD couples to the PD. In the case of the L361R with W434F mutations, any interruption of the chain eliminated the Q-V and G-V crossing; and in the case of the L366H with W434F mutations, any interruption of the chain restored the inactivation of the W434F mutation [Citation36]. Altogether, these observations (1) suggest that the chained amino acid residues are functionally coupled among them and (2) demonstrate that the proposed residues are indeed the molecular basis for the noncanonical coupling mechanism. It is important to point out that the noncanonical mechanism uncovered by our group may not be the only one. It is quite possible that other paths are present as suggested by other researchers’ groups [Citation38,Citation39,Citation48].
We extended our studies of gating currents without the W434F mutation that is in the selectivity filter. Instead, we eliminated K+ currents with the V478W mutation that prevents the activation gate to populate the open conformation [Citation49]. We used the generalized interaction-energy analysis to estimate the free energy of activation by the median voltage of the Q-V curves [Citation50,Citation51] and found that the residues forming the noncanonical mechanism interact. Indicating that the noncanonical coupling operates without the W434F mutation. We used double mutant cycle analysis to show the interaction energy between residues S411 and F433. During gating events, these two residues interact with a net energy of 0.5 kcal/mol (2 kcal/mol per channel when accounting for four VSDs). Assuming that this interaction represents most of the energy exchanged between the VSD and the PD apposed together, this result suggests that most likely this noncanonical mechanism operates at a level of energy that would not be enough to gate the PD, a molecular event that requires 14 kcal/mol per channel [Citation50]. However, this energy should be accounted for both canonical and the noncanonical coupling process. Nevertheless, we found that the noncanonical coupling was efficient in transferring information to both the activation gate and to the selectivity filter gate [Citation36]. The modulation of the activation gate by means of the noncanonical coupling mechanism is undoubtedly more relevant when some mutations are present in the VSD and in the PD [Citation45]. The detailed mechanism by which the activation gate is modulated by this novel coupling mechanism remains elusive. On the other hand, the selectivity filter gate is indeed important for the channel’s C-type (slow) inactivation as we showed that this novel mechanism directly couples VSD to the selectivity filter in the PD [Citation36,Citation37]. It is important to notice that C-type inactivation in VGKC is central for cell excitability as the number of C-type-inactivated channels determines the action potential firing frequency in neurons.
The interface between VSD and PD emerges as a new target site for VGKC modulation as we showed it to be active in the VSD-to-PD overall coupling mechanism. Indeed, it is quite possible that this interface and the noncanonical mechanism play a role in the normal function of ether-a-go-go (EAG) K+ channels family (KV10.1 and KV11.1), VGKC that exhibit non-swapping domain architecture. These channels may operate without the classical canonical coupling covalent (S4-S5 linker disrupted), suggesting the presence of a noncanonical coupling mechanism [Citation52]. Moreover, the Kv7.1 channel has been reported to be open by VSD movements from the intermediate and from the activated states [Citation53,Citation54], and it has been suggested that a noncanonical coupling underlies the coupling between VSD and the activated open state [Citation55], demonstrating the relevance of this mechanism in the VGKC family.
Even though progress has been made on the understanding of the molecular mechanism involved in the noncanonical coupling, more structural and biophysical analyses are needed for a full understanding of its operation. Biophysical, computational, and structural assays should further contribute to explain the underling properties of this novel coupling mechanism to the function of the VGKC.
4- How ligand- and voltage-domain communicate in HCN channels (Andrea Saponaro and Anna Moroni)
Hyperpolarization-activated cyclic nucleotide-gated (HCN) channels are the molecular correlate of If (or Ih) current, which plays a key role in controlling the rhythmic activity in cardiac pacemaker cells and spontaneously firing neurons [Citation56]. HCN channels are cationic non-selective channels and allow the passage of a depolarizing mixed Na+ and K+ current. In humans, the HCN channel family comprises four members (HCN1-HCN4, about 50% sequence identity with each other) [Citation57,Citation58]. HCN channels are activated by hyperpolarization and further modulated by the direct binding of the second messenger cAMP to their C-terminal CNBD. Binding of cAMP facilitates voltage-dependent opening of the channel, that occurs at more positive values. Thus, it seems clear that the transmembrane voltage sensor domain (VSD) and the cytosolic cyclic nucleotide binding domain (CNBD) are functionally coupled and influence each other, although the molecular determinant of their connection was not assigned with certainty till recently.
Cyclic AMP-induced conformational changes in the CNBD propagate, through the C-linker, to the transmembrane core (hereafter cAMP pathway), where it modulates channel voltage-dependency. The cAMP pathway within the cytoplasmic region have been extensively characterized at the level of the CNBD [Citation59–61] and within the C-linker region [Citation62–68].
By contrast, the molecular details of the propagation of the cAMP signal from the cytoplasmic region to the transmembrane core are less clear. The C-linker is directly connected to the S6 helix that form the main gate at the intracellular pore entrance, the so-called bundle crossing [Citation69]. It is generally believed that the cAMP-induced rotation of the C-linkers acts directly on the S6, modulating the bundle crossing [Citation68,Citation70]. This simple model does not take into account that, in HCN channels, cAMP is ineffective in the absence of hyperpolarization, indicating that the VSD blocks the C-linker movement in depolarization and supporting the view of a direct connection between the VSD and the C-linker that bypasses the pore. More recently, a direct connection between the VSD and the cAMP pathway has been proposed based on functional studies [Citation71,Citation72]. In these studies, ligand binding and channel activation were simultaneously measured. Kinetic analysis of channel activation and cAMP binding indicated the existence of a reciprocal influence between the VSD and the ligand binding machinery. In the proposed model, the movement of the VSD following hyperpolarizing voltages increases the binding affinity in the CNBD, and, concomitantly, cAMP binding to the CNBD facilitates SVD movements. Thus, activation by voltage and cAMP binding are intimately coupled and influence each other. Kusch and co-workers have also shown that the increase in affinity of the CNBD precedes pore opening, reinforcing previous hypothesis that the coupling mechanism between VSD and C-linker/CNBD bypasses the pore. These results were further enriched by an independent study [Citation72], indicating that coupling of the two mechanisms occurs during channel deactivation as well. In silico analysis performed on HCN channels by reduced mechanical model [Citation73] or by Molecular dynamics simulation [Citation74] indicate that the cAMP-induced movements of the C-linker are efficiently coupled with and propagated to the VSD. Taken together, these results demonstrated that in HCN channels the VSD and the ligand binding machinery are tightly connected and functionally coupled.
Initial attempts to elucidate a possible interaction between the S4-S5 linker and the C-linker in mouse HCN2 [Citation75,Citation76], were biased by the structure of a Kv channel, the only 3D model for voltage gated ion channels present at that time. This caused the misleading attribution of a critical connection between S5 and S6 helices of HCN pore to the S4-S5 linker – C-linker pair. Possible contacts were further investigated by cross-linking cytosolic and transmembrane domains in spHCN by means of high-affinity metal bridges or disulfide bridges introduced in the protein by means of point mutations. In this way, it was possible not only to show that the S4-S5 linker and the C-linker are functionally coupled but that the relative orientation of the S4-S5 linker and the C-linker changes during channel gating [Citation77,Citation78]. It is nonetheless worth noting that though these studies suggested that the S4–S5 linker and the C-linker are in close proximity during the entire process of channel opening, they do not necessarily indicate the existence of physical contacts between them.
Only recently, the cryo-EM structures of HCN1 and HCN4 allowed the identification of the molecular contacts between the C-linker and the VSD.
The structure of human HCN1 [Citation69] revealed the presence of the N-terminal HCN domain (HCNd), a soluble domain that wedges itself between the VSD and the C-linker. Its role is to bridge these two domains by means of a network of molecular contacts that are crucially needed for cAMP response. A recent study demonstrates by functional and theoretical experiments that the C-linker rotation induced by cAMP binding, is transmitted to the VSD by means of the HCNd. This mechanism operates in HCN1, HCN2 and HCN4 and is therefore a general feature of HCNs [Citation79].
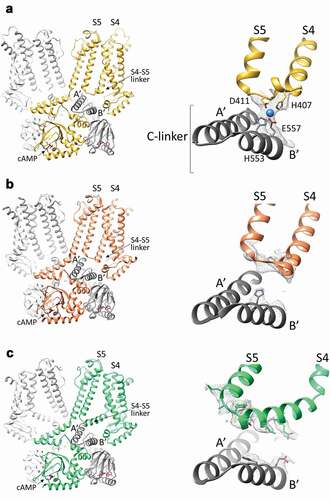
An additional mechanism for the transmission of the cAMP signal to the VSD, emerged from the structure of rabbit HCN4. The HCN4 structure solved in the presence of cAMP demonstrated, for the first time, the existence of a physical connection between the S4–S5 linker and the underlying C-linker. Specifically, four residues, H407 and D411 from the S4-S5 linker, and H553 and E557 from the C-linker, generates a sort of tetrahedral arrangement, defined as the ‘tetrad’ that coordinates a magnesium ion (). Of note, such a metal ion binding site is also present in human HCN4 (density map file EMD-0094, PDB ID 6GYO). Functional data indicated that the tetrad promotes the cAMP-dependent coupling between the cytoplasmic and the transmembrane domains and controls the cAMP response in HCN4 channels. It is, however, worth noting that the effect of either the disruption of the tetrad via mutations of the residues forming it, or the removal of Mg2+, only halved the cAMP response of HCN4 channels. This confirms that the main mechanism for cAMP transmission to the VSD in HCN4 is the one mediated by the HCNd and that the tetrad is an additional pathway employed to propagate the cAMP signal in HCN4 only. Indeed, HCN1 doesn’t show contacts between the S4–S5 linker and the C-linker, neither when the channel is in the resting state () [Citation69], nor when it is chemically stabilized in its voltage activated-like conformation ()) [Citation69]. This is due to the fact that, despite sequence identity, the S4-S5 linker in HCN1 is one helix turn shorter than in HCN4, preventing tetrad formation ()). Also, even though the structure of HCN2 is not yet available, the results of functional studies suggest that the tetrad does not control cAMP response in this isoform as well [Citation80,Citation81].
Figure 4. The S4-S5 linker in HCN4 and HCN1 channels. A, Left, structure of the cAMP-bound HCN4 channel tetramer, in a cross-membrane view. For clarity, only two subunits in the tetramer are shown in full (yellow and light grey), while for the other two subunits (dark and light grey), only the cytosolic C-linker/CNBD domains are presented. The transmembrane S4 and S5 helices, the S4-S5 linker connecting them and the first two helices (A’ and B’) of the cytoplasmic C-linker are labelled. Right, ribbon representation of the S4-S5 linker of one subunit (yellow) and the underlying C-linker (A’-B’ helices) of the adjacent subunit (grey) of HCN4. Four residues forming the ion coordination site (tetrad) in HCN4 are shown as sticks and labelled (H407, D411, H553, and E557). The ion coordinated by the tetrad is represented as a blue sphere. The density map for the region of the tetrad is shown as grey mesh. B, Left, structure of the cAMP-bound HCN1 channel tetramer in the resting state (PDB: 5U6P), in a cross-membrane view. For clarity, only two subunits in the tetramer are shown in full (orange and light grey), while for the other two subunits (dark and light grey) only the cytosolic C-linker/CNBD domains are presented. The transmembrane S4 and S5 helices, the S4-S5 linker connecting them and the first two helices (A’ and B’) of the cytoplasmic C-linker are labelled. Right, ribbon representation of the S4-S5 linker of one subunit (orange) and the underlying C-linker (A’-B’ helices) of the adjacent subunit (grey) of HCN1 in the resting state. Shown as grey mesh is the density map for the S4-S5 linker and of the corresponding residues of the HCN4 tetrad located in B’ helix of C-linker. Only the two corresponding histidines forming the ion tetrad HCN4 are shown as sticks as for its side chain there is density. C, Left, structure of the cAMP-bound HCN1 channel tetramer in the activated-like state (PDB: 6UQ), in a cross-membrane view. For clarity, only two subunits in the tetramer are shown in full (green and light grey), while for the other two subunits (dark and light grey) only the cytosolic C-linker/CNBD domains are presented. The transmembrane S4 and S5 helices, the S4-S5 linker connecting them and the first two helices (A’ and B’) of the cytoplasmic C-linker are labelled. Right, ribbon representation of the S4-S5 linker of one subunit (green) and the underlying C-linker (A’-B’ helices) of the adjacent subunit (grey) of HCN1 in the activated-like state. Shown as grey mesh is the density map for the S4-S5 linker and of the corresponding residues of the HCN4 tetrad located in B’ helix of C-linker. The residues of these regions for whose side chains there is density are shown as sticks
Interactions between the S4–S5 linker and the C-linker were identified and functionally proven to be involved in the gating of KAT1 [Citation82] and CNG channels [Citation83,Citation84], both closely related to HCN channels. A more distantly related channel, the voltage and Ca2+ activated BK, also employ magnesium ion to coordinate the association between the VSD and the cytosolic ligand binding machinery [Citation81].
In summary, the cytosolic and the transmembrane domains of HCN channels are physically and functionally connected in different ways. One modality of connection, which is shared by all isoforms, involves the HCNd that bridges the C-linker to the VSD. A second modality, which is HCN4-specific, connects the C-linker to the S4-S5 linker through the tetrad, a magnesium ion coordination site.
Disclosure statement
The authors declare no conflict of interest.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
References
- González C, Baez-Nieto D, Valencia I, et al. K(+) channels: function-structural overview. Compr Physiol. 2012;2:2087–24.
- Doyle DA, Morais Cabral J, Pfuetzner RA, et al. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280:69–77.
- LeMasurier M, Heginbotham L, Miller C. KcsA: it’s a potassium channel. J Gen Physiol. 2001;118:303–314.
- Morais-Cabral JH, Zhou Y, MacKinnon R. Energetic optimization of ion conduction rate by the K+ selectivity filter. Nature. 2001;414:37–42.
- Zhou Y, MacKinnon R. The occupancy of ions in the K+ selectivity filter: charge balance and coupling of ion binding to a protein conformational change underlie high conduction rates. J Mol Biol. 2003;333:965–975.
- Begenisich T, De Weer P. Potassium flux ratio in voltage-clamped squid giant axons. J Gen Physiol. 1980;76:83–98.
- Hodgkin AL, Keynes RD. The potassium permeability of a giant nerve fibre. J Physiol. 1955;128:61–88.
- Neyton J, Miller C. Discrete Ba2+ block as a probe of ion occupancy and pore structure in the high-conductance Ca2+ -activated K+ channel. J Gen Physiol. 1988;92:569–586.
- Alcayaga C, Cecchi X, Alvarez O, et al. Streaming potential measurements in Ca2+-activated K+ channels from skeletal and smooth muscle. Coupling of ion and water fluxes. Biophys J. 1989;55:367–371.
- Köpfer DA, Song C, Gruene T, et al. Ion permeation in K+ channels occurs by direct Coulomb knock-on. Science. 2014;346:352–355.
- Langan PS, Vandavasi VG, Weiss KL, et al. Anomalous X-ray diffraction studies of ion transport in K. Nat Commun. 2018;9:4540.
- Öster C, Hendriks K, Kopec W, et al. The conduction pathway of potassium channels is water free under physiological conditions. Sci Adv. 2019;5. DOI:10.1126/sciadv.aaw6756.
- Hille B. Ionic channels of excitable membranes (3rd ed.). 2021, Sunderland, MA: Sinauer Associates Inc.
- Nelson PH. A permeation theory for single-file ion channels: one- and two-step models. J Chem Phys. 2011;134:165102.
- Cooper KE, Gates PY, Eisenberg RS. Diffusion theory and discrete rate constants in ion permeation. J Membr Biol. 1988a;106:95–105.
- Szabo A. Theory of polarized fluorescent emission in uniaxial liquid crystals. J Chem Phys. 1980;72:4620–4626.
- Bernetti M, Masetti M, Rocchia W, et al. Kinetics of Drug Binding and Residence Time. Annu Rev Phys Chem. 2019;70:143–171.
- Bernèche S, Roux B. Energetics of ion conduction through the K+ channel. Nature. 2001;414:73–77.
- Bullerjahn JT, von Bülow S, Hummer G. Optimal estimates of self-diffusion coefficients from molecular dynamics simulations. J Chem Phys. 2020;153:024116.
- Bernèche S, Roux B. A microscopic view of ion conduction through the K+ channel. Proc Natl Acad Sci U S A. 2003;100:8644–8648.
- Hilder TA, Corry B, Chung SH. Multi-ion versus single-ion conduction mechanisms can yield current rectification in biological ion channels. J Biol Phys. 2014;40:109–119.
- Hartel AJW, Shekar S, Ong P, et al. High bandwidth approaches in nanopore and ion channel recordings - A tutorial review. Anal Chim Acta. 2019;1061:13–27.
- Roux B. Ion Conduction and Selectivity in K +Channels. Annu Rev Biophys Biomol Struct. 2005;34:153–171.
- Swartz KJ. Towards a structural view of gating in potassium channels. Nat Rev Neurosci. 2004;5:905–916.
- Rauh O, Hansen UP, Mach S, et al. Extended beta distributions open the access to fast gating in bilayer experiments-assigning the voltage-dependent gating to the selectivity filter. FEBS Lett. 2017a;591:3850–3860.
- Rauh O, Urban M, Henkes LM, et al. Identification of Intrahelical Bifurcated H-Bonds as a New Type of Gate in K(+) Channels. J Am Chem Soc. 2017b;139:7494–7503.
- Thiel G, Baumeister D, Schroeder I, et al. Minimal art: or why small viral K(+) channels are good tools for understanding basic structure and function relations. Biochim Biophys Acta. 2011;1808:580–588.
- Cooper KE, Gates PY, Eisenberg RS. Surmounting barriers in ionic channels. Q Rev Biophys. 1988b;21:331–364.
- Cordero-Morales JF, Jogini V, Chakrapani S, et al. A multipoint hydrogen-bond network underlying KcsA C-type inactivation. Biophys J. 2011;100:2387–2393.
- Lacroix JJ, Hyde HC, Campos FV, et al. Moving gating charges through the gating pore in a Kv channel voltage sensor. Proc Natl Acad Sci U S A. 2014;111:E1950–1959.
- Starace DM, Bezanilla F. A proton pore in a potassium channel voltage sensor reveals a focused electric field. Nature. 2004;427:548–553.
- Campos FV, Chanda B, Roux B, et al. Two atomic constraints unambiguously position the S4 segment relative to S1 and S2 segments in the closed state of Shaker K channel. Proc Natl Acad Sci U S A. 2007;104:7904–7909.
- Vargas E, Bezanilla F, Roux B. In search of a consensus model of the resting state of a voltage-sensing domain. Neuron. 2011;72:713–720.
- Kalstrup T, Blunck R. S4–S5 linker movement during activation and inactivation in voltage-gated K + channels. Proc Natl Acad Sci U S A. 2018;115:E6751–e6759.
- Long SB, Campbell EB, Mackinnon R. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science. 2005a;309:897–903.
- Bassetto CA, Carvalho-de-Souza JL, Bezanilla F. Molecular basis for functional connectivity between the voltage sensor and the selectivity filter gate in Shaker K(+) channels. Elife. 2021. DOI:10.7554/eLife.63077
- Carvalho-de-Souza JL, Bezanilla F. Noncanonical mechanism of voltage sensor coupling to pore revealed by tandem dimers of Shaker. Nat Commun. 2019;10:3584.
- Fernández-Mariño AI, Harpole TJ, Oelstrom K, et al. Gating interaction maps reveal a noncanonical electromechanical coupling mode in the Shaker K(+) channel. Nat Struct Mol Biol. 2018;25:320–326.
- Lee SY, Banerjee A, MacKinnon R. Two separate interfaces between the voltage sensor and pore are required for the function of voltage-dependent K(+) channels. PLoS Biol. 2009;7:e47.
- Chen X, Wang Q, Ni F, et al. Structure of the full-length Shaker potassium channel Kv1.2 by normal-mode-based X-ray crystallographic refinement. Proc Natl Acad Sci U S A. 2010;107:11352–11357.
- Long SB, Campbell EB, Mackinnon R. Voltage sensor of Kv1.2: structural basis of electromechanical coupling. Science. 2005b;309:903–908.
- Sun J, MacKinnon R. Cryo-EM Structure of a KCNQ1/CaM Complex Reveals Insights into Congenital Long QT Syndrome. Cell. 2017;169:1042–1050.e1049.
- Wang W, MacKinnon R. Cryo-EM Structure of the Open Human Ether-à-go-go-Related K(+) Channel hERG. Cell. 2017;169:422–430.e410.
- Whicher JR, MacKinnon R. Structure of the voltage-gated K+ channel Eag1 reveals an alternative voltage sensing mechanism. Science. 2016;353:664–669.
- Carvalho-de-Souza JL, Bezanilla F. Nonsensing residues in S3-S4 linker’s C terminus affect the voltage sensor set point in K(+) channels. J Gen Physiol. 2018;150:307–321.
- Perozo E, MacKinnon R, Bezanilla F, et al. Gating currents from a nonconducting mutant reveal open-closed conformations in Shaker K+ channels. Neuron. 1993;11:353–358.
- Yang Y, Yan Y, Sigworth FJ. How does the W434F mutation block current in Shaker potassium channels? J Gen Physiol. 1997;109:779–789.
- Conti L, Renhorn J, Gabrielsson A, et al. Reciprocal voltage sensor-to-pore coupling leads to potassium channel C-type inactivation. Sci Rep. 2016;6:27562.
- Kitaguchi T, Sukhareva M, Swartz KJ. Stabilizing the closed S6 gate in the Shaker Kv channel through modification of a hydrophobic seal. J Gen Physiol. 2004;124:319–332.
- Chowdhury S, Chanda B. Estimating the voltage-dependent free energy change of ion channels using the median voltage for activation. J Gen Physiol. 2012;139:3–17.
- Chowdhury S, Haehnel BM, Chanda B. A self-consistent approach for determining pairwise interactions that underlie channel activation. J Gen Physiol. 2014;144:441–455.
- Lörinczi É, Gómez-Posada JC, de la Peña P, et al. Voltage-dependent gating of KCNH potassium channels lacking a covalent link between voltage-sensing and pore domains. Nat Commun. 2015;6:6672.
- Cui J. Voltage-Dependent Gating: novel Insights from KCNQ1 Channels. Biophys J. 2016;110:14–25.
- Zaydman MA, Kasimova MA, McFarland K, et al. Domain-domain interactions determine the gating, permeation, pharmacology, and subunit modulation of the IKs ion channel. Elife. 2014;3:e03606.
- Hou P, Eldstrom J, Shi J, et al. Inactivation of KCNQ1 potassium channels reveals dynamic coupling between voltage sensing and pore opening. Nat Commun. 2017;8:1730.
- Robinson RB, Siegelbaum SA. Hyperpolarization-activated cation currents: from molecules to physiological function. Annu Rev Physiol. 2003;65:453–480.
- Biel M, Wahl-Schott C, Michalakis S, et al. Hyperpolarization-activated cation channels: from genes to function. Physiol Rev. 2009;89:847–885.
- Wahl-Schott C, Biel M. HCN channels: structure, cellular regulation and physiological function. Cell Mol Life Sci. 2009;66:470–494.
- Akimoto M, Zhang Z, Boulton S, et al. A mechanism for the auto-inhibition of hyperpolarization-activated cyclic nucleotide-gated (HCN) channel opening and its relief by cAMP. J Biol Chem. 2014;289:22205–22220.
- Puljung MC, DeBerg HA, Zagotta WN, et al. Double electron-electron resonance reveals cAMP-induced conformational change in HCN channels. Proc Natl Acad Sci U S A. 2014;111:9816–9821.
- Saponaro A, Matzapetakis M, Moroni A, et al. Structural rearrangements occurring on HCN2 CNBD domain upon cAMP binding. European Biophysics Journal With Biophysics Letters. 2013;42:S181.
- Craven KB, Olivier NB, Zagotta WN. C-terminal movement during gating in cyclic nucleotide-modulated channels. J Biol Chem. 2008;283:14728–14738.
- Craven KB, Zagotta WN. Salt bridges and gating in the COOH-terminal region of HCN2 and CNGA1 channels. J Gen Physiol. 2004;124:663–677.
- Lolicato M, Bucchi A, Arrigoni C, et al. Cyclic dinucleotides bind the C-linker of HCN4 to control channel cAMP responsiveness. Nat Chem Biol. 2014;10:457–462.
- Lolicato M, Nardini M, Gazzarrini S, et al. Tetramerization dynamics of C-terminal domain underlies isoform-specific cAMP gating in hyperpolarization-activated cyclic nucleotide-gated channels. J Biol Chem. 2011;286:44811–44820.
- VanSchouwen B, Melacini G. Role of Dimers in the cAMP-Dependent Activation of Hyperpolarization-Activated Cyclic-Nucleotide-Modulated (HCN) Ion Channels. J Phys Chem B. 2018;122:2177–2190.
- Xu X, Vysotskaya ZV, Liu Q, et al. Structural basis for the cAMP-dependent gating in the human HCN4 channel. J Biol Chem. 2010;285:37082–37091.
- Zagotta WN, Olivier NB, Black KD, et al. Structural basis for modulation and agonist specificity of HCN pacemaker channels. Nature. 2003;425:200–205.
- Lee CH, MacKinnon R. Structures of the Human HCN1 Hyperpolarization-Activated Channel. Cell. 2017;168:111–120.e111.
- Wainger BJ, DeGennaro M, Santoro B, et al. Molecular mechanism of cAMP modulation of HCN pacemaker channels. Nature. 2001;411:805–810.
- Kusch J, Biskup C, Thon S, et al. Interdependence of receptor activation and ligand binding in HCN2 pacemaker channels. Neuron. 2010;67:75–85.
- Wu S, Vysotskaya ZV, Xu X, et al. State-dependent cAMP binding to functioning HCN channels studied by patch-clamp fluorometry. Biophys J. 2011;100:1226–1232.
- Weißgraeber S, Saponaro A, Thiel G, et al. A reduced mechanical model for cAMP-modulated gating in HCN channels. Sci Rep. 2017;7:40168.
- Gross C, Saponaro A, Santoro B, et al. Mechanical transduction of cytoplasmic-to-transmembrane-domain movements in a hyperpolarization-activated cyclic nucleotide-gated cation channel. J Biol Chem. 2018;293:12908–12918.
- Chen J, Mitcheson JS, Tristani-Firouzi M, et al. The S4-S5 linker couples voltage sensing and activation of pacemaker channels. Proc Natl Acad Sci U S A. 2001;98:11277–11282.
- Decher N, Chen J, Sanguinetti MC. Voltage-dependent gating of hyperpolarization-activated, cyclic nucleotide-gated pacemaker channels: molecular coupling between the S4-S5 and C-linkers. J Biol Chem. 2004;279:13859–13865.
- Kwan DC, Prole DL, Yellen G. Structural changes during HCN channel gating defined by high affinity metal bridges. J Gen Physiol. 2012;140:279–291.
- Prole DL, Yellen G. Reversal of HCN channel voltage dependence via bridging of the S4-S5 linker and Post-S6. J Gen Physiol. 2006;128:273–282.
- Porro A, Saponaro A, Gasparri F, et al. The HCN domain couples voltage gating and cAMP response in hyperpolarization-activated cyclic nucleotide-gated channels. Elife. 2019;8. DOI:10.7554/eLife.49672
- Saponaro A, Bauer D, Giese MH, et al. Gating movements and ion permeation in HCN4 pacemaker channels. Mol Cell. 2021;81:2929–2943.e6.
- Yang H, Hu L, Shi J, et al. Mg2+ mediates interaction between the voltage sensor and cytosolic domain to activate BK channels. Proc Natl Acad Sci U S A. 2007;104:18270–18275.
- Clark MD, Contreras GF, Shen R, et al. Electromechanical coupling in the hyperpolarization-activated K(+) channel KAT1. Nature. 2020;583:145–149.
- Li M, Zhou X, Wang S, et al. Structure of a eukaryotic cyclic-nucleotide-gated channel. Nature. 2017;542:60–65.
- Zheng X, Fu Z, Su D, et al. Mechanism of ligand activation of a eukaryotic cyclic nucleotide-gated channel. Nat Struct Mol Biol. 2020;27:625–634.