
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Gas phase experiments combined with ultrafast technologies can provide information on the intrinsic properties of molecular systems at picosecond, femtosecond, or even attosecond timescales. However, these experiments are often limited to relatively simple model systems. In this context, electrospray ionization sources (ESI) have offered new perspectives as they allow to produce large or fragile molecular ions in the gas phase, mimicking molecules in their natural environment. While time-resolved UV-visible ultrafast experiments on molecular ions have been successfully developed over the past decades, efforts are still required to perform experiments using ultrashort extreme ultraviolet (XUV) pulses with the goal of reaching attosecond resolution. In this article, we present recent results obtained using the combination of ultrafast technologies and ESI sources. We show that ultrafast dynamics experiments can be performed on molecular ions without ion trapping devices and can reveal UV-induced charge transfer in small peptides with controlled micro-environment. Non-adiabatic relaxation dynamics in large (bio)molecular ions is also presented. Such experiments are compatible with high harmonic generation XUV sources as shown here in the case of a metal complex. These ultrafast dynamics studies on large molecular ions offer new perspectives in attosecond science.
Graphical abstract
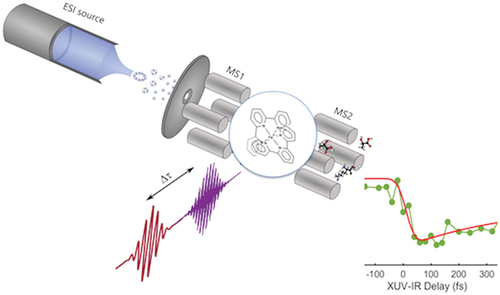
1. Introduction
Many photoinduced processes are encountered in nature and technology, spanning from photosynthesis or DNA photoprotection to photovoltaics devices [Citation1]. They usually correspond to an absorption of light followed by ultrafast dynamics, whose final product provides insight on specific properties. As a consequence, the complete understanding or optimization of these processes requires the knowledge of the dynamical behavior and pathways, from the absorption to the final product, of the photo-induced reaction. Previous works have revealed numerous mechanisms occurring after light absorption at ultrashort timescales, such as charge transfer, vibronic relaxation, intramolecular vibrational redistribution, spin dynamics, structural rearrangements, or interaction with the surrounding environment or surfaces [Citation2]. These processes enable the fine understanding of natural phenomena and show the intricated role of the intrinsic properties of molecules and of their natural environment. In this context, studying isolated neutral molecules provided a powerful approach to define intrinsic properties. However, most molecules in their natural environment carry a charge, which has a strong influence on their electronic structure and global properties. Consequently, the study of neutral gas-phase molecules is of interest for the fundamental understanding of photo-physics and photochemistry, but it generally less relevant in regard to natural processes. Additionally, biologically relevant species are often fragile, therefore molecular beam experiments are not suitable. Electrospray Ionization techniques (ESI) have allowed to produce such fragile molecular ions in the gas phase [Citation3]. Because ESI makes ‘elephant fly’ as described by the Nobel Prize winner J.B. Fenn [Citation4], the possibility to investigate very large and complex isolated systems offered new perspectives in a wide range of applications from analytical chemistry to fundamental photophysics and photochemistry. Recent ultrafast experiments have been developed in that context using ion traps to study photoinduced dynamics following ultraviolet (UV) light excitation [Citation5].
At the same time, the development of ultrafast light sources evolved drastically with the emergence of secondary sources based on high harmonic generation (HHG). This provided new perspectives in time-resolved experiments by allowing to study processes induced by high energy photons and by reaching femtosecond and even attosecond time resolution [Citation6]. With a rapid development in the field, experiments have revealed photoionization time delays in atoms [Citation7] and molecules [Citation8], hole migration in small peptides [Citation9], dynamics in amino-acids [Citation10], non-adiabatic dynamics in polycyclic aromatic hydrocarbons (PAH) [Citation11]. Yet, all investigations in the gas phase have focused on neutral atoms and molecules [Citation12]. However, ions are not only crucial for biologically relevant investigations but offer new questions to address because the charge carried by the molecule will strongly modify correlation effects and charge dynamics. Moreover, the charge state may originate from various factors, such as a lack or an excess of electrons or protons or the presence of a counter-ion, which strongly influences the molecular properties.
Here, we discuss current efforts to address ultrafast dynamics in molecular ions. We present recent UV-IR pump-probe experiments that reveal ultrafast charge transfer dynamics in peptide ions without using any trapping devices. We show that the charge transfer dynamics can be tuned by controlling the microenvironment of the molecule. We also present an extension of this work, unraveling non-adiabatic relaxation in more complex ions: the reserpine molecules. We finally present the first results using HHG XUV-IR pump-probe experiments performed in a metal-complex molecule. This work opens new opportunities for the development of ultrafast XUV attosecond experiments in chemically relevant species.
2. Experimental set-up for ‘on the fly’ time-resolved experiments in molecular ions
Our experimental setup consists of an electrospray ionization source combined with a triple quadrupole mass spectrometer instrument and a pump-probe scheme (see ). The ions are produced by the ESI source and mass selected by a first quadrupole (MS1). They are then transferred to the interaction region where they are submitted to laser irradiation in a pump and probe pulse sequence. Finally, the photo-products are analyzed in a second quadrupole (MS2). The interaction is characterized by monitoring the ionization and fragmentation yield as a function of the pump-probe time delays that is measured.
Figure 1. Experimental setup for XUV/UV-IR pump-probe experiment in molecular ions. The experiment combines an HHG XUV source (generation chamber), an XUV-IR interferometer (recombination chamber), an ESI source and a triple quadrupole mass spectrometer. For UV experiments the generation and recombination chambers are simply replaced by third harmonic generation (THG) crystals to reach 266 nm (see text for a detailed description).
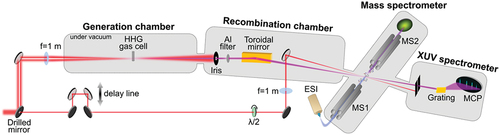
We used the output of a 25fs, 800 nm, 2 mJ, 5 kHz amplified femtosecond laser system. The laser beam is split into two parts: the pump arm and the probe arm. The pump arm is used to produce a pulse either in the UV (267 nm, 80fs, produced by third-harmonic generation) or XUV (30 eV, 20fs, produced by HHG). In the case of the UV light, the UV pump beam passes through a reflective delay line, before being recombined with the IR probe beam. Using a lens with f = 1 m focal length, both pump and probe beams are focused on the ion beam after MS1. Finally, the products of the interaction are analyzed by MS2. For the XUV pulse, the femtosecond 800 nm laser beam of the pump arm is focused into a gas cell to generate XUV light through HHG process using rare gas atoms such as Xenon, Krypton, and Argon. This allows generating photon energies in the range of 10 to 50 eV. After the HHG source, the residual IR is removed using a metallic Al filter that transmits only harmonics above the 9th one and overall sculpts the XUV spectrum. A toroidal mirror is used to focus the XUV beam at the exit of MS1. The second part of the IR beam crossed a delay line (refractive line made of two wedges) that is used to control the time delay between the XUV and the IR pulse. The delayed beam is focused and recombined with the XUV pulse using a drilled mirror in the MS1. Here also the products of the interaction is measured by MS2. In a typical pump-probe experiment, the mass distribution is recorded for different XUV-IR time delays.
In this experiment, we make use of the 5 kHz repetition rate of the laser that offers a good signal-to-noise ratio without using an ion trap (i.e, ‘on-the-fly’ configuration). The gas-phase ions travel in the spectrometer at a typical speed of few 100s m/s, thus ensuring that the ions travel several centimeters between two pump-probe sequences (200 µs), which is much larger than the focus size (150–200 µm). It means that compared to other existing configurations that use an ion trap, our experimental set-up ensures that molecular ions interact only once with the pump and probe pulses.
In the following, the pump-probe scans consist of recording the mass spectrum obtained with MS2 at each pump-probe time delay. The pump-probe signal corresponds to the integration of the yield of a given ion measured by MS2, as a function of the time delay. A fitting procedure was used, using a typical model:
where τ is the lifetime in the exponential decay, t0 the time delay, Astep the amplitude difference between the yield at negative and positive time delays (it corresponds to the long time delay asymptotic step observed in the experimental signals at positive time delays), and θ is the Heaviside function.
3. UV and XUV induced ultrafast dynamics in molecular ions
In the following, we present the results of a time-resolved UV-IR experiment performed on small peptides [Citation13]. In this experiment, we demonstrate how our set-up can be used to monitor the influence of the microenvironment on the ultrafast charge transfer dynamics in biomolecular ions. As an extension of this work, we also present other recent results where charge and energy dynamics in UV or XUV excited complex molecular ions are investigated. In the case of UV excitation, the general process involves the excitation of a chromophore that carries a localized electronic excited state. The excited population is not stationary and evolves from one electronic state to another, inducing a charge transfer and/or a vibrational heating of the molecule. When the XUV pulses are used, the main relaxation channel following excitation is the photoionization. Photoionization leads to a molecule with an additional positive charge and a substantial electronic and vibrational energy. As a consequence, as in the case of neutral molecules, charge and energy transfer occur following the XUV ionisation.
3.1 UV induced dynamics in molecular ions
3.1.1. Charge transfer in UV excited peptides
We first consider the case of positively charged peptides. Peptides are chains of amino acids linked by peptide bonds forming biological polymers (proteins) that have crucial functions in living organisms. In their natural environment peptides can be either positively or negatively charged depending on the solvent pH. We study the photoactive properties of positively charged species under UV excitation. The results presented here have been discussed in a recent publication [Citation12]. The concept of the experiment is to investigate how modifications of the atomic environment of a biomolecule can influence its non-stationary properties. The versatility of the ESI-MS setup allows to form and isolate complex fragile systems which is illustrated here by the possibility to investigate tryptophan and tryptophan-containing molecules (Tryptophan: C11H12N2O2, M = 204.2 g/mol; Alanyl-tryptophan: C14H17N3O3, M = 275.3 g/mol) complexed with an atomic ion (H+ or Na+ in our experiment). Because this simple modification of the environment of the molecule might have an impact on its electronic structure and therefore on its ultrafast properties, we have performed time-resolved experiments to study its impact on ππ*-πσ* dynamics.
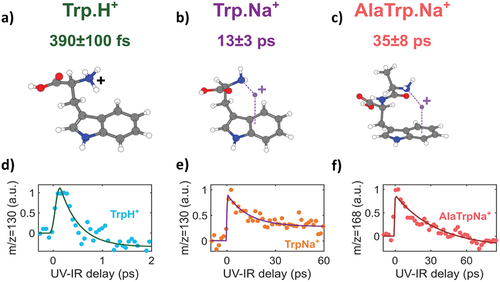
The couplings between ππ* and πσ* states have an important role in photochemical properties [Citation14]. In the case of protonated tryptophan, the ππ* state is excited by the UV pulse and the subsequent dynamics is investigated by measuring the variations of the fragmentation yield of the m/z = 130 channel as a function of the UV pump- IR probe time delay. The m/z = 130 fragment corresponds to dissociation of the protonated tryptophan molecule after localization of the positive charge on the chromophore. Therefore, it is a good signature of the positive charge transfer from the charge carrier (H+) to the indole ring (electron transfer from the indole to the proton). To understand the process at play, one should notice that the UV pump pulse populates the ππ* state, and that the IR probe pulse further excites the population to higher electronic states, eventually leading to fragments such as m/z = 130. As a consequence, the time-dependent signal of the fragment is a signature of the population dynamics in the ππ* state. The results of the measurements are presented in . The time delay-dependent signal can be fitted with a single exponential model defined by a time constant. The time constant is interpreted as the characteristic time of the coupling between the ππ* state and the lower-lying πσ* state, which is responsible for the transfer of an electron from the indole to the added proton. Note that a step is observed on the long time delay. This step is strongly influenced by the technical limitation in terms of long-term stability of the experiment. As a consequence, no physical meaning can be inferred from the amplitude of this step but it is taken into account in the analysis (see fitting formula). We studied this charge transfer dynamics in three tryptophan-containing systems: protonated tryptophan (TrpH+) (), the sodiated tryptophan (TrpNa+) () and the sodiated alanyl-tryptophan (AlaTrpNa+) ()). In each case, the dynamics is probed by using a fragment that is a signature of the charge transfer such as m/z = 130 in the case of TrpH+. The fitting procedure yielded a time constant τ = 390 ± 100 fs () in the case of TrpH+, τ = 13 3 ps for TrpNa+ () and τ = 35 ± 8 ps for AlaTrpNa+ (). The extracted time constant becomes higher and higher: it increases by a factor 30 when going from TrpH+ to TrpNa+ and by a factor of 100 when going to AlaTrpNa+. We note that this variation is induced by a simple change of the local environment of the tryptophan chromophore (either an added charged atom or molecular group).
Figure 2. 267 nm-800 nm pump-probe experiment on charged peptides. (a,b,c) Structure of the 3 peptides of interest where a modification of the environment of the chromophore (change in the adduct atom and change of the molecular chain added to the chromophore) induces a variation of the dynamics. Time-dependent ion signal for: (d) fragment m/z = 130 in TrpH+, (e) fragment m/z = 130 in TrpNa+, (f) fragment m/z = 168 in AlaTrpNa+.
We proposed a simple model to rationalize this observation. The observed dynamics corresponds to the ππ*-πσ* coupling leading to a population transfer from the localized ππ* state to the πσ* state. The transfer time constant τπ*-σ* depends on the energy gap between the two states. This simple energy gap law shows that the increase of the time constant is a direct consequence of the increase in the energy gap between the coupled states, and that it is induced by the changes in the micro-environment. This influence is understood as a tuning of the relative energy gap between the ππ* and the πσ* states due to electrostatic forces. These forces can be split in 2 contributions: the global and the local stabilization effects. As discussed in ref [Citation15] the ππ* state is delocalized over the chromophore and is therefore sensitive to the global interaction with the charged moiety. The πσ* state is localized around the N-X+ bond (where X is H or Na), making the state more sensitive to the local electrostatic interaction with the charge carrier. The global interaction depends mainly on the affinity of the tryptophan group to the considered charge carrier (proton affinity or sodium ion affinity in our case). The local effect is mainly determined by the coulomb attraction between the NH2 moiety and the adduct (which depends on the distance between the NH2 and the atom). Overall, the two effects combine and determine the lifetime of the electron dynamics in the molecule. This shows that the dynamics can be tuned by controlling the environment of the chromophore. More complex structures could be investigated using our experiment, also using different modifications of the environment.
3.1.2. Non-adiabatic relaxation in UV excited protonated reserpine
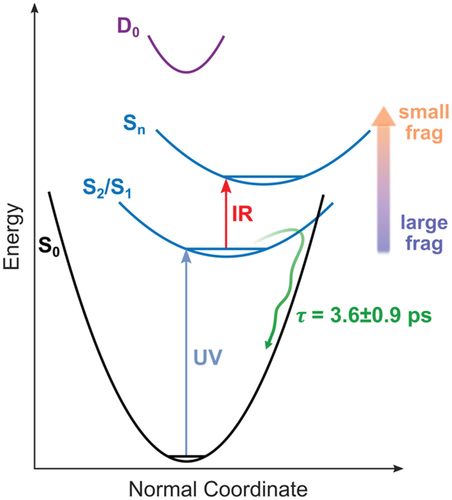
In this second example, we considered a slightly more complex object, the protonated reserpine molecule. The reserpine molecule (C33H40N2O9, M = 608.7 g/mol) is an alkaloid used in the treatment of high blood pressure. The structure of the molecule is presented in . It contains 2 aromatic rings that can efficiently absorb UV light, which are connected by a molecular bridge. We consider the protonated form of the molecule where the proton is located near the N atom of the bridge between the two aromatic rings. We used the ultrashort 266 nm UV laser pulse to excite the molecule, followed by a time delayed 800 nm IR probe pulse. The 266 nm pulse energy is maintained at low energy per pulse (5 µJ) and the IR at 840 µJ (intensity of about 1–2.1012 W.cm−2). The mass spectrum obtained by combining the two laser pulses is presented in . One notices that the same fragments are observed in the case of UV-only experiments (see ref [Citation15]). The result of the pump-probe experiment is shown in . It corresponds to the time-dependent variation of the fragmentation yield measured by monitoring the channels m/z = 381, 395, 436, and 448, renormalized by the UV-only ion signal. These signals show an exponential decay after an initial population in the case of m/z = 381, 395 (), and depletion is observed for m/z = 436 and 448 (). We note that the signal for higher masses is depleted while the lower ones are populated. Apart from the population or depletion character, the fitting procedure shows that a single time constant is sufficient to represent all the transient signals presented. The fitting procedure leads to a time constant of 3.6 ± 0.9 ps. The only difference observed between the various fragments is the intensity of the asymptotic step at long time delays, which depends on the considered fragment. The interpretation of this time constant is presented in . The UV excitation occurs either on the methoxy indole or trimethoxybenzoyl aromatic chromophores. It leads to the population of first electronic excited states that relax to the ground state in approximately 4 ps through a conical intersection. This relaxation corresponds to the transfer of electronic energy to vibrational energy. Because the UV excitation is localized on the aromatic rings, the first step of the decay process must involve local modes. After complete intramolecular vibrational redistribution (IVR), the molecule subsequently fragments, leading to large fragments such as m/z 436 and 448. When the molecule interacts with the combination of the UV and IR pulse, the population of the electronic excited state is depleted and further excited to higher electronic states, leading to the appearance of small fragments such as m/z 381 and 395. Depending on when the IR excitation occurs along the relaxation dynamics, the depletion is less and less efficient. This is what is observed in where the fragment yield of m/z 381 and 395 is increased around zero delay and progressively decreases due to the non-adiabatic dynamics (the contrary for the larger fragments). We conclude that the lifetime of the photoactive first electronic states in protonated reserpine is determined by an efficient non-adiabatic relaxation through conical intersections. This shows that the reserpine molecule efficiently converts UV excitation and electronic energy into internal vibrational heating, which has a direct impact on the fluorescence yield and could explain the fluorescence quenching observed in the molecule [Citation16].
Figure 3. 267 nm-800 nm pump-probe experiment performed on protonated reserpine. The molecular structure is presented on the top. a) Mass spectrum obtained at the temporal overlap of the 267 nm and 800 nm pulses. b) Time-dependent ion signal for: b) fragment m/z = 381, c) fragment m/z = 395, d) fragment m/z = 436, and e) fragment m/z = 448 (blue dots). A fit with a decay function is also shown for all the fragment (blue curve), giving an overall time constant of 3.6 ± 0.9 ps.
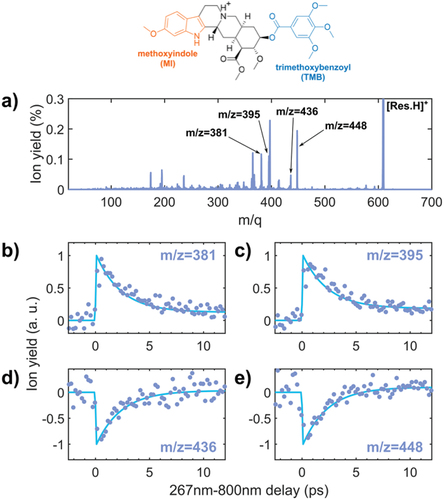
Figure 4. Schematic representation of the processes at play in protonated reserpine. UV excitation to S2/S1 occurs, inducing a relaxation down to the S0 state within 3.6 ps that can eventually lead to large fragments. The dynamics is probed by the 800 nm pulse that excites higher energy electronic states. This leads to the depletion of the channels inducing large fragments and the population to small fragments channels.
3.2. XUV-induced ultrafast dynamics in molecular ions
The previous experiments have been developed using UV light as a pump. We note that these experiments have provided time-resolved results without any use of a trapping device. Ion traps are often used to increase the density of molecules with the drawback of limiting the spectrometric approaches that can be applied to these ions. Here, despite the low ion density, it is possible to obtain high-quality time-resolved signals. Recent developments in ultrafast science concern the use of XUV ultrashort pulses to investigate processes down to the attosecond timescale [Citation17]. Increasing the complexity of the systems and studying molecules that are relevant to other research field is of crucial importance. The study of molecular ions with ultrashort XUV pulses enters in this category. We have recently developed experiments in that direction by combining the flexibility of the ESI sources in terms of sample manipulation and the HHG sources in terms of versatility of the production of XUV radiation. This first ever experimental set up combining ESI and HHG source has been presented elsewhere (see ref [Citation18]). Time-dependent experiments using ultrashort XUV pulses is of prime interest to investigate ionization-induced dynamics. While alternative methods using multiphoton transitions can also be used, single-photon ionization remains a more direct approach in which the ejection of deep valence electrons is an efficient process with strong correlation effects [Citation19]. Following the ionization, the creation of an additional charge on the molecule can induce charge or structural rearrangements and destabilize the molecular structure. In that context, the study of molecular ions is essential because the intrinsic charge of the system allows to modify electronic interactions in the molecule. The femtosecond resolution provided by HHG sources offers the opportunity to identify the first steps of this mechanism. In the following, we present the first report of the ultrafast dynamics following XUV ionization in molecular ions. We have performed XUV pump- IR probe experiments on bis(terpyridine) iron(II) metal complex ion, Fe(terpy)22+ (, C30H22N6Fe2+, M = 522.4 g/mol). This system has been widely studied in the past to investigate fundamental aspects of solar cells properties [Citation20].
Figure 5. XUV-IR pump-probe experiment in the bis(terpyridine) Iron(II) ion. The structure of the molecule shows the double pyridine-based structures surrounding the Fe atom. The pump-probe signal obtained in XUV-IR experiments is based on the variation of the m/z 174 fragment yield. (Experimental data (green dots), red curve (fit)). It can be interpreted as the charge transfer and structural changes induced by the removal of an electron from the metal complex.
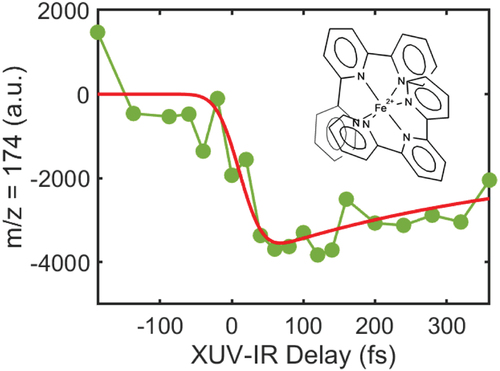
In the experiment, the XUV spectrum is centered around 30 eV and the IR pulse intensity is maintained around 1012 W/cm [Citation2]. We measured the time delay-dependent variation of the fragmentation yield for the channel m/z 174. This fragment corresponds to the ionized metal complex ion Fe(terpy)23+. This signal shows an ultrafast dynamics on a few 100 fs timescales. One can speculate that the dynamics is related to the metal-ligand interaction. In our experiment, the XUV radiation excites the pyridine molecules leading to the removal of an electron from the aromatic rings. As a consequence, electron transfer from the Fe2+ to the pyridine might occur to neutralize the charge. Therefore, the measured time constant could be related to the charge transfer and first molecular rearrangements prior to fragmentation. Although the experiment is still exploratory, it shows the feasibility and interest of performing XUV induced ultrafast dynamics in ionic metal complexes. With the wide tuneability of HHG sources, the intrinsic properties of metal complexes could be investigated in detail, for instance using the resonances of the Fe atoms in the XUV domain.
4. Conclusion
Ultrafast dynamics in molecular ions is a subject of broad interest with applications ranging from fundamental physics to biochemistry. First, it allows to investigate the influence of additional charges on the out of equilibrium molecular properties. It also allows an explicit connection between fundamental physical-chemistry and questions of biological or chemical interest. This is due to the fact that ions are closer to the molecular composition in their natural liquid-phase environments. Here, we have presented new time-resolved experiments where molecular ions can be studied with no trapping device. This will allow us to consider the implementation of complex spectroscopic techniques such as photoelectron spectroscopy. As demonstrated in a previous article [Citation21], the combination of velocity map imaging spectrometry and ESI on-the-fly configuration offers the possibility to have access to angularly resolved electron spectra. Implementing this configuration with time-resolved scheme would be of high interest. We have shown that this approach allowed us to study micro-environment effects on UV induced charge transfer in small peptides, and to measure non-adiabatic relaxation in complex ions to understand excited states lifetimes and optical properties of complex molecules. We have also shown that the study of XUV-induced ultrafast dynamics in molecular ions has become possible. This is a completely unexplored field and could serve at revealing the effect of the charge on hole dynamics, proton motion, energy relaxation, which have been recently reported in neutral model systems. Such experiments could also provide information on the first instants on the production of photoinduced radicals in proteins and DNA, which play a role in radiation damage. Efforts in that direction are pursued in our group. Finally, these results pave the way to the development of attosecond chemistry in complex molecular structures.
Acknowledgments
This work was supported by the CNRS, ANR Circé ANR-16-CE30-0012, FAUST ANR-21-CE30-0052-01, and COST Action CA18222 “Attosecond Chemistry” (AttoChem).
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Domcke W, Yarkony DR. Role of conical intersections in molecular spectroscopy and photoinduced chemical dynamics. Annu Rev Phys Chem. 2012;63:325–14.
- May V, Kühn O. Charge and energy transfer dynamics in molecular systems. Hoboken, New Jersey, U.S.: John Wiley & Sons; 2011.
- Yamashita M, Fenn JB. Electrospray ion source. Another variation on the free-jet theme. J Phys Chem. 1984;88:4451.
- Fenn JB. Electrospray wings for molecular elephants (Nobel lecture). Angew Chem. 2003;42:3871–3894.
- Soorkia S, Jouvet C, Grégoire G. UV photoinduced dynamics of conformer-resolved aromatic peptides. Chem Rev. 2020;120:3296–3327.
- Krausz F, Ivanov M. Attosecond physics. Rev Mod Phys. 2009;81:163.
- Schultze M, Fieß M, Karpowicz N, et al. Delay in photoemission. Science. 2010;328:1658.
- Nandi S, Plésiat, E, Zhong, S, et al . Attosecond timing of electron emission from a molecular shape resonance. Sci Adv. 2020;6:eaba7762.
- Calegari F, Ayuso D, Trabattoni A, et al. Ultrafast electron dynamics in phenylalanine initiated by attosecond pulses. Science. 2014;346:336–339.
- Lara-Astiaso M, Galli M, Trabattoni A, et al. Attosecond pump–probe spectroscopy of charge dynamics in tryptophan. J Phys Chem Lett. 2018;9:4570–4577.
- Hervé M, Despré V, Castellanos Nash P, et al. Ultrafast dynamics of correlation bands following XUV molecular photoionization. Nat Phys. 2021;17:327.
- Lépine F, Ivanov MY, Vrakking MJJ. Attosecond molecular dynamics: fact or fiction? Nat Photonics. 2014;8:195–204.
- Hervé M, Boyer A, Brédy R, et al. Controlled ultrafast ππ*-πσ* dynamics in tryptophan-based peptides with tailored micro-environment. Communications Chemistry. 2021;4:124.
- Sobolewski ALG, Domcke W, Dedonder-Lardeux C, et al. Excited-state hydrogen detachment and hydrogen transfer driven by repulsive 1πσ* states: a new paradigm for nonradiative decay in aromatic biomolecules. Phys Chem Chem Phys. 2002;4:1093–1100.
- Brédy R, Hervé M, Boyer A, et al. Non-ergodic fragmentation of protonated reserpine using femtosecond laser activation. Int J Mass Spectrom. 2022;471:116729.
- Sakal EH, Merrill EJ. J Pharm Assoc. 1954;43:709.
- Nisoli M, Decleva P, Calegari F, et al. Attosecond electron dynamics in molecules. Chem Rev. 2017;117:10760–10825.
- Hervé M, Boyer A, Brédy R, et al. On-the-fly investigation of XUV excited large molecular ions using a high harmonic generation light source. Sci Rep. 2022;12:13191.
- Marciniak A, Despré V, Loriot V, et al. Electron correlation driven non-adiabatic relaxation in molecules excited by an ultrashort extreme ultraviolet pulse. Nature Commun. 2019;10:337.
- Han FS, Higuchi M, Kurth DG. Metallosupramolecular polyelectrolytes self-assembled from various pyridine ring-substituted bisterpyridines and metal ions: photophysical, electrochemical, and electrochromic properties. J Am Chem Soc. 2008;130:2073–2208.
- Papalazarou E, Cauchy C, Barillot T, et al. Combined electrospray ionization source with a velocity map imaging spectrometer for studying large gas phase molecular ions. Analyst. 2012;137:3496–3501.