Abstract
We conducted a biogeographic analysis based on a dense phylogenetic hypothesis for the early branches of corvids, to assess geographic origin of the New World jay (NWJ) clade. We produced a multilocus phylogeny from sequences of three nuclear introns and three mitochondrial genes and included at least one species from each NWJ genus and 29 species representing the rest of the five corvid subfamilies in the analysis. We used the S-DIVA, S-DEC, and BBM analyses implemented in RASP to create biogeographic reconstructions, and BEAST to estimate timing of NWJ diversification. Biogeographic reconstructions indicated that NWJs originated from an ancestor in the Eastern Palearctic or Eastern + Western Palearctic, diversified in Mesoamerica and spread subsequently to North and South America; the group has been diversifying in the New World since the late Miocene.
Introduction
The New World jays (NWJs) are a monophyletic group of corvids [Citation1,2], presently considered to comprise seven genera (Aphelocoma, Calocitta, Cyanocitta, Cyanocorax, Cyanolyca, Gymnorhinus and Psilorhinus); the ~36 species [Citation3,4] represent the product of a radiation across much of the Americas [Citation1]. The group has long been a focus of research in behavioral ecology, in light of complex behavioral repertoires, particularly as regards social behavior [Citation5–7].
The origins of corvids can be traced to Australia, from where the ancestor of the family dispersed to Asia, followed by radiations in Asia, Europe and elsewhere [Citation8]. Ericson and colleagues [Citation9–11] provided evidence that the origin of the oscine passerines, to which the Corvidae belongs, dates to the split of the Australo-Papuan tectonic plate from Antarctica in the early Tertiary, ~53M years ago. Thus, the replacement of the early Tertiary rainforests in Australia by drier vegetation may have placed presumably forest-adapted early corvids in more open habitats, resulting in a new radiation that led to the present global distribution of the family. However, a recent study [Citation12] estimated the age of the family at ~20M years. Colonization of the New World by corvids occurred more recently, apparently via a trans-Beringian route [Citation8]. The ancestor of NWJs, thought to be jay-like lineages related to Cissa and Urocissa, reached North America 10−8M years ago, in the Miocene [Citation13]. Once in the Americas, a rapid radiation in South America by the early Pliocene, was apparently followed by a secondary radiation in North America [Citation1]. This hypothesis was not supported by the later analyses of Bonaccorso and Peterson [Citation14], who offered support for a hypothesis that the NWJ ancestor was in Mesoamerica or North America + Mesoamerica and radiated into North and South America independently. The origins of this diverse clade are of considerable interest in light of questions about the biogeography of Asian and American tropical groups [Citation15], morphological innovation [Citation2] and the evolutionary origins of social behavior [Citation2].
Several previous studies have examined the evolutionary history, phylogeny and biogeography of the NWJs, but invariably based on sparse representation of NWJ genera and limited outgroup sampling. De los Monteros and Cracraft [Citation1] proposed a first hypothesis of intergeneric relationships in the group based on cytochrome b sequences. Ericson et al. [Citation11] added markers but included only single species per genus. Saunders and Edwards [Citation16] examined mitochondrial DNA control region (CR) variation among representatives of all NWJ genera. Finally, Bonaccorso and Peterson [Citation14] derived a multilocus phylogeny with much-improved NWJ species representation and included a historical biogeographic analysis that concluded that NWJ origins were in Mesoamerica or Mesoamerica + North America.
These previous studies, however, did not provide sufficient detail in terms of representation of taxa of NWJs and other corvids to permit thorough understanding the geographic origin of NWJs or to identify their closest corvid relatives. The key deficiency has been in terms of representation of taxa from the deepest branches of the corvid phylogeny; thus, we here derive a denser phylogenetic hypothesis by deriving DNA sequences for these early lineages, placing several species of treepies, blue magpies and green magpies in a molecular phylogeny for the first time, to permit more robust biogeographic analyses. In light of this new data set, we discuss geographic origins of NWJs and offer a new hypothesis.
Methods
Taxon and gene sampling
Taxon sampling included at least one species from each NWJ genus, 29 species representing the rest of the five corvid subfamilies and Lanius as a definitive outgroup (see Appendix 1 for a summary). Frozen tissue samples (muscle tissue) were obtained from the American Museum of Natural History, Louisiana State University Museum of Natural Science, National Taiwan Normal University and the University of Kansas Natural History Museum. We also added DNA sequences from GenBank from previous studies [Citation1,11,14,17–34] that overlapped our sequenced gene regions.
DNA sequences were obtained for three mitochondrial genes (cytochrome b, or Cytb; nicotinamide adenine dinucleotide dehydrogenase subunit 2, or ND2; and nicotinamide adenine dinucleotide dehydrogenase subunit 3, or ND3), and three nuclear introns (the seventh intron of the beta-fibrinogen gene, or βfib7; the second intron of the myoglobin gene, or Myo2; and the fifth intron of the transforming growth factor β2, or TGFβ2). Genes were amplified using the following primers: Cytb, L14851 [Citation35] and L428 and H494 [Citation36]; ND2, L5215 and H6313 [Citation37]; ND3, L10755 and H11151 [Citation38]; βfib7, FIB-B17U and FIB-B17L [Citation39]; Myo2, Myo2 [Citation40] and Myo3f [Citation41]; and TGFβ2, TGF5 and TGF6 [Citation35].
DNA extraction, PCR amplification, sequencing, alignment and model selection
DNA was extracted from frozen tissues using proteinase K digestion under manufacturer’s protocols (Qiagen DNeasy tissue kit). PCR amplifications were performed using Promega GoTaq Flexi DNA polymerase. We employed a touchdown protocol for the amplification of ND2 and ND3, with annealing temperatures of 55 and 50°C, respectively. Constant annealing temperatures of 58, 50, 59 and 56°C were used for Cytb, βfib7, Myo2 and TGFβ2, respectively. PCR products were sent to Beckman Coulter Genomics (Danvers, MA) for purification and final sequencing.
Sequences were cleaned, assembled and aligned in Geneious 8.1.6 (Biomatters, Auckland, New Zealand) and checked against an automated alignment in MUSCLE [Citation42]. Sequence data were partitioned by codon position for mitochondrial DNA and by gene for nuclear introns. We used MrModeltest 2.3 [Citation43] to identify appropriate substitutional models for each partition under the Akaike information criterion (AIC).
Phylogenetic analysis
Single-gene matrices were concatenated using Geneious 8.1.6 to produce an overall mitochondrial data set, a nuclear intron data set and a total combined matrix. Phylogenies were estimated with Bayesian analysis (BA) and maximum-likelihood (ML) inference methods. GARLI v2.01 [Citation44] was used for the maximum-likelihood analysis. We performed 100 independent analyses and chose the tree with the best likelihood score. Nodal support values for topologies were evaluated via 500 nonparametric bootstrap replicates [Citation45]. The 50% majority-rule consensus tree was acquired in PAUP* 4.0 [Citation46]; 70% bootstrap support was considered as indicative of solid nodal support [Citation47,48].
Bayesian phylogenetic estimates were obtained in MrBayes 3.2.2 [Citation49–51]. Two independent Markov chain Monte Carlo (MCMC) runs of 2 107 generations using four chains (temp = 0.1) per run were conducted. We sampled trees every 2000 generations and discarded the first 25% of the trees as burn-in. We used TRACER 1.6 [Citation52] to assess convergence of parameter estimates and posterior probabilities. The remaining trees were summarized to obtain a 50% majority-rule consensus tree.
Ancestral area reconstructions
We employed S-DIVA (Statistical Dispersal-Vicariance Analysis), ‘Bayes−Lagrange’ or Statistical Dispersal-Extinction-Cladogenesis (S-DEC [Citation53,54]), and Bayesian Binary Method (BBM) analysis implemented in RASP 3.2 [Citation55,56] to reconstruct ancestral areas of corvid lineages. DIVA is an event-based method in which vicariance is assumed and includes potential contributions for dispersal and extinction by assigning a cost [Citation57]. S-DIVA is an expansion of Bayes-DIVA implemented by Nylander et al. [Citation58] that incorporates the entire posterior distribution of tree topologies, thus accounting for both phylogenetic and ancestral state uncertainty [Citation55,56]. S-DEC model has been implemented using the source code for the C++ version of Lagrange [Citation59]. This model gives the likelihood of all possible biogeographic scenarios estimated at a given node and summarizes biogeographic reconstructions across all user-supplied trees [Citation56]. BBM is a statistical procedure in which a full hierarchical Bayesian approach is used to reconstruct ancestral distributions at ancestral nodes [Citation60]; it is implemented in RASP [Citation56], wherein the source code of MrBayes 3.1.2 is used to create the MCMC analysis.
We defined 8 geographic areas (A = North America, B = Mesoamerica, C = South America, D = Western Palearctic, E = Eastern Palearctic, F = Afrotropics, G = Indo-Malaya, H = Australia). We assigned each species included in the phylogenetic analysis to a region or regions, based on published range maps [Citation61]. S-DIVA, S-DEC and BBM analyses were conducted using 10,000 postburn-in trees derived from the overall combined data matrix from the Bayesian Markov chain Monte Carlo (MCMC) procedure implemented in BEAST 1.8.2 [Citation62]. For the S-DEC analysis, we considered the probability of dispersal between areas as equal, and all values in the dispersal constraint matrix were set to 1. BBM analysis used the maximum clade probability tree (condensed tree) from the BEAST analysis and included two independent runs of 10 chains (temp = 0.1) that ran for 1M generations, sampling every 100 generations, but discarding the initial 25% as burn-in. We used the F81 + G evolutionary model for the analysis, based on the results of the model testing described above. In all analyses, the maximum number of areas included in ancestral distributions was set to 2. We excluded the broadly distributed outgroup Lanius from these analyses.
Timing of diversification
The fossil record for passerine birds is scarce and many described fossils lack information necessary for use as informative calibration points [Citation63]. Thus, we estimated the time of diversification of NWJs using previously published ND2 substitution rate priors using BEAST 1.8.2 under a relaxed clock model. We unlinked nucleotide substitution models, and used MrModelTest 2.3 [Citation43] to obtain best-fitting substitutional models. We linked clock and tree models, used a birth-death speciation process and ran them for 5 × 108 generations, sampling every 50,000th generation. Convergence was examined in TRACER [Citation52]; all effective sample size (ESS) values were > 200. We discarded the initial 25% of samples as burn-in. Calibrating a phylogeny based on empirically determined mitochondrial DNA substitution rates offers only a rough estimate of actual time, though lacking fossils within the clade, such an external rate calibration offers the only option for linking to actual dates [Citation64]. ND2 is widely studied in ornithology [Citation65]; we used two empirically estimated substitution rate priors derived from ND2 substitution rates (2.4 and 3.3%/lineage/million years) for Hawaiian honeycreepers [Citation66] that were derived based on three recently obtained geologic calibration points, to estimate divergence times for various corvid lineages.
Results
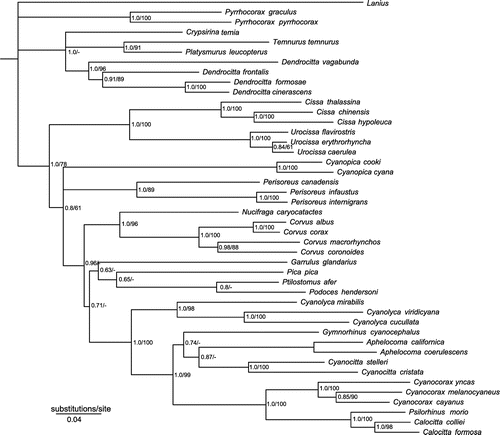
Topologies inferred via ML and BA analysis were highly congruent for the concatenated mitochondrial and nuclear sequence datasets (Figure 1; also see Appendix 2 for concatenated mitochondrial, and nuclear datasets). Results from the BEAST analysis were similar to the ML and BA topologies, except that Pyrrhocorax was placed as sister to clade A in the BEAST analysis (posterior probability 0.84); ML and BA could not resolve the placement of this genus. In general, these mostly congruent phylogenetic hypotheses all agreed in identifying five clades within the corvids: A, including Temnurus, Platysmurus, Crypsirina, and Dendrocitta; B, including Cissa and Urocissa; C, including Cyanopica and Perisoreus; D, including Pica, Podoces, Ptilostomus, Garrulus, Nucifraga and Corvus; and E, including Cyanolyca, Aphelocoma, Gymnorhinus, Cyanocitta, Cyanocorax, Psilorhinus and Calocitta. The NWJ clade (clade E) was placed as sister to clade D with strong support (posterior probability 1.0, 100% ML bootstraps). Monophyly of the clades C, D and E was highly supported (1.0, 87%), although the sister relationship of Perisoreus and Cyanopica was ambiguous (0.72, 47%). Placement of Podoces as sister to Ptilostomus [Citation11] was not well-supported; in contrast, a sister relationship between Podoces and Pica was better-supported, with a posterior probability of 0.95% and 58% bootstrap support.
S-DIVA, S-DEC and BBM ancestral area reconstructions for NWJs produced congruent results, estimating Mesoamerica as the most probable ancestral area for the common ancestor of clade E (Figure ). According to S-DIVA analysis, Mesoamerica + Eastern Palearctic was the most probable ancestral area (p = 1.0) for the common ancestor of clades D and E. S-DEC also showed Mesoamerica + Eastern Palearctic as the most probable ancestral area (p = 0.63), and Eastern Palearctic was the other possible area (p = 0.37). BBM results were equivocal, with four possible ancestral areas: Eastern Palearctic (p = 0.40), Eastern Palearctic + Western Palearctic (p = 0.31), Western Palearctic (p = 0.16) and ambiguous (p = 0.13). Because all NWJ lineages were connected to American areas, uncertainty in phylogenetic arrangements within clade C had no effect on our reconstructions of areas of lineages ancestral to the NWJs.
BEAST relaxed-clock analysis based on the two substitution rate priors indicated that the time to the most recent common ancestor for NWJs was 5.8−4.3M years. The split of the NWJ clade (clade E) from its sister clade (clade D) was estimated at 6.7−5.0M years (Figure ). Our analysis estimated that the Corvidae likely originated in the late Miocene, 8.0−5.9M years ago.
Discussion
Our biogeographic analysis was based on denser taxonomic sampling of the early branches of the corvid phylogeny than had been available in any previous study. Our results provided solid evidence regarding the geographic origin of NWJs. Mesoamerica + Eastern Palearctic or Eastern Palearctic was consistently identified as the most probable ancestral area for the ancestor of the NWJ and its sister clade; thus, the lineage probably reached the New World from the Eastern Palearctic through a trans-Beringian route. An interesting observation that resulted from the ancestral area reconstruction was the absence of the common ancestor of clades D and E in North America (Figure ). A plausible explanation to this observation can be drawn from studies on other taxa: for example, Fox [Citation67], based on mammalian fossils, documented aridification of North America during the late Miocene, thus linking it with increased seasonality. Similarly, Kohn and Fremd [Citation68] documented decreased faunal diversity in western North America in the late Miocene, which they postulated to coincide with increased aridity and seasonality. The ancestor of clades D and E probably largely bypassed North America during the late Miocene, perhaps using western coastal habitats under the climate-stabilizing effects of the Pacific Ocean that may have offered temporary corridors for passage from the Eastern Palearctic to Mesoamerica. Hence, early cladogenesis—at least among extant taxa—of the NWJ lineage probably took place either in Mesoamerica or in the Eastern Palearctic.
Our results indicated that the NWJs are most closely related to a clade comprising Pica, Podoces, Garrulus and others. Ericson et al. [Citation11] identified Indo-Malaya as the ancestral area of origin of these taxa. In another study, De los Monteros and Cracraft [Citation1] recovered Perisoreus as the sister taxon to the NWJ clade. Additionally, Craig et al. [Citation13] described a close relationship between the NWJ ancestor and Cissa + Urocissa. Our results refute these hypotheses, with better taxonomic representation, a more robust multilocus data-set, and better support (Figure ).
Figure 1. Phylogeny of Corvidae based on Bayesian and Maximum Likelihood analyses of the combined data-set. Numbers at the nodes refer to Bayesian posterior probability/ML bootstrap support. Asterisks indicate 1.0 posterior probability and 100% bootstrap support. Letters at nodes indicate five major clades of the family.
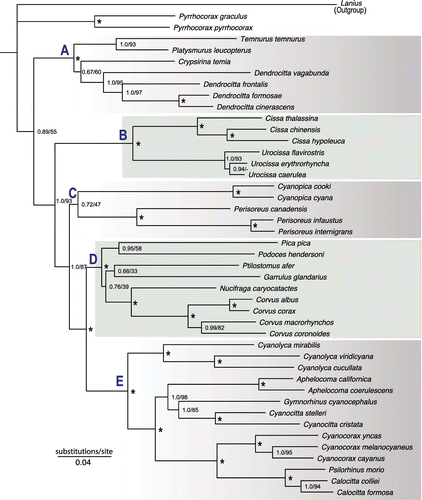
Figure 2. Reconstruction of the ancestral area of lineages of Corvidae. Pie charts indicate probability of various ancestral area combinations. Pie charts at nodes were derived from S-DEC analysis; the smaller charts above and below at the nodes obtained from S-DIVA and BBM analyses, respectively. Ancestral areas are shown only for basal nodes. The New World jay clade (clade E), its sister clade (clade D) and all other basal clades (C, B and A) of corvids, are outlined in light blue, orange and green boxes, respectively.
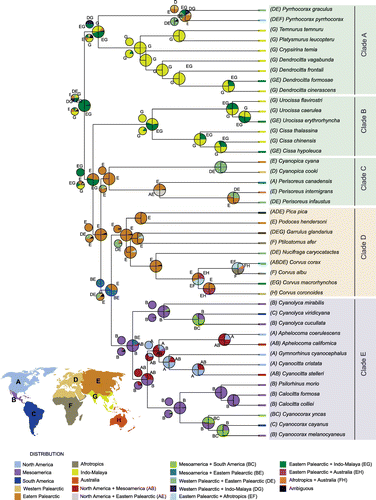
All of our analyses identified Mesoamerica as the most probable area for the most recent common ancestor of NWJs. This result narrows down the results of Bonaccorso and Peterson [Citation14], which showed the ancestral distribution of the NWJs as either Mesoamerica or Mesoamerica + North America. The proposed origin of Calocitta + Cyanocorax + Psilorhinus in Mesoamerica, dispersing thence into South America [Citation14], was supported in our analysis. However, our results were unclear as to whether the Aphelocoma + Cyanocittta + Gymnorhinus clade originated in North America vs. North America + Mesoamerica.
De los Monteros and Cracraft [Citation1] proposed the idea that the origin of the NWJs was the result of a single dispersal event into the Americas, which could have taken place around the early Miocene. However, our time calibration estimates indicated a later (Miocene−early Pliocene) origin for the NWJs (Figure ), in which the ancestor probably dispersed to the New World from the Eastern Palearctic, or Eastern + Western Palearctic, around 6.7−5.0 M years ago (Figure ). This event could have taken place when a continuous coniferous belt linked Northern Asia and America across Beringia from mid-to-late Miocene to the late Pliocene [Citation69], yet the timing of our reconstructions does not appear to coincide with any specific geological events that might have triggered the dispersal event.
Figure 3. Approximate timeframe for the diversification of the Corvidae, with particular focus on New World jays. Time scales are based on ND2 substitution rates of 2.4% and 3.3% per lineage per million years. Bars at nodes indicate the highest posterior density (HPD) interval. Numbers at nodes refer to Bayesian posterior probabilities. Letters at nodes indicate the five major clades of the family.
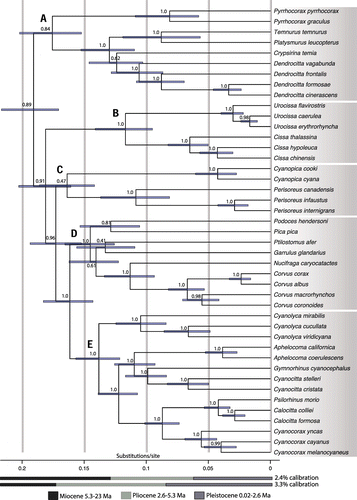
Our time calibration estimated a late Miocene‒early Pliocene origin for the most recent common ancestor (MRCA) of NWJs. This timing makes intuitive sense considering the absence of the MRCA in North America given the continent’s increased seasonality and aridity by that time period, as stated above [Citation67]. The time calibration further estimated the age of the family Corvidae at ~8.0−5.9 M years, substantially younger than the recent estimate of ~20M years in the phylogeny of corvoid passerine birds [Citation12]. However, we note that the timing of Corvidae–Laniidae split of the recent phylogeny of songbirds based on ~5000 loci by Moyle et al. [Citation70] coincides with our estimate for the same node (not shown here). Moreover, our time estimates for the MRCA of the South American genera (2.8−2.0M years for Cyanolyca, and 2.3−1.7M years for Cyanocorax) make intuitive sense, since dispersal of theses clades from Mesoamerica into South America should postdate the emergence of the Isthmus of Panama, around 2.8 M years ago [Citation71]. However, we emphasize that our time estimates are rough estimates of actual time and must be considered with caution.
Our results provide novel insights into the biogeographic origin of NWJs, offering a solid hypothesis based on different ancestral area reconstruction methods, backed by strong support from a robust, multilocus phylogeny. However, clearly, more complete taxon representation with genome-level sampling could perhaps improve support for some remaining ambiguous nodes in the phylogenetic hypothesis. Such an improved phylogeny would help to resolve ambiguities associated with some of our phylogenetic and geographic hypotheses, and could allow insight into other important questions, such as tropical-temperate transitions in the family.
Associate Editor: Elisa Bonaccorso
Disclosure Statement
No potential conflict of interest was reported by the authors.
Acknowledgments
We are grateful to the museum collectors and field workers who collected specimens used in this study. We thank the collections managers and curators of American Museum of Natural History, Louisiana State University Museum of Natural Science, National Taiwan Normal University, and the University of Kansas Natural History Museum, for providing tissue samples. For providing sequences of Crypsirina temia, Dendrocitta cinerascens, and Platysmurus leucopterus, we thank Carl Oliveros. We thank Matthew Girard for helping with data cleaning and statistical analysis. For wise guidance in the laboratory, we thank Alana Alexander. Thilina De Silva provided support in laboratory work and statistical analysis. We are grateful to Leo Smith for the assistance and guidance in the lab. The Office of the Provost, University of Kansas, supported this study financially.
Related Research Data
References
- De los Monteros AE, Cracraft J. Intergeneric relationships of the New World jays inferred from cytochrome b gene sequences. Condor. 1997;99:490–502.
- Zusi RL. A feeding adaptation of the jaw articulation in New World jays (Corvidae). Auk. 1987;104:665–680.
- Goodwin D. Crows of the world. Ithaca (NY): Cornell University Press; 1976.
- Madge S, Burn H. Crows and jays: A guide to the crows, jays and magpie of the world. Boston: Houghton Mifflin Company; 1994.
- Brown JL. Alternate routes to sociality in jays—with a theory for the evolution of altruism and communal breeding. Am Zool. 1974;14:63–80.10.1093/icb/14.1.63
- Brown JL. Social organization and behavior of the Mexican Jay. Condor. 1963;65:l26–l53.
- Peterson A. Phylogeny and rates of molecular evolution in the jays of the genus Aphelocoma (Corvidae). Auk. 1992;109:134–148.
- Sibley CG, Ahlquist J. The phylogeny and classification of the Australo-Papuan Passerine birds. Emu. 1985;85:1–14.10.1071/MU9850001
- Ericson PG, Christidis L, Cooper A, et al. A Gondwanan origin of passerine birds supported by DNA sequences of the endemic New Zealand wrens. Proc R Soc Lond B. 2002;269:235–241.10.1098/rspb.2001.1877
- Ericson PG, Johansson US. Phylogeny of Passerida (Aves: Passeriformes) based on nuclear and mitochondrial sequence data. Mol Phylogenet Evol. 2003;29:126–138.10.1016/S1055-7903(03)00067-8
- Ericson PG, Jansén AL, Johansson US, et al. Inter-generic relationships of the crows, jays, magpies and allied groups (Aves: Corvidae) based on nucleotide sequence data. J Avian Biol. 2005;36:222–234.10.1111/jav.2005.36.issue-3
- Jønsson KA, Fabre P-H, Kennedy JD, et al. A supermatrix phylogeny of corvoid passerine birds (Aves: Corvides). Mol Phylogenet Evol. 2016;94:87–94.10.1016/j.ympev.2015.08.020
- Craig A, Feare C, Hoyo Jd, et al. Handbook of the birds of the world. Vol. 14: Bush-shrikes to Old World sparrows. Barcelona: Lynx Edicions; 2009.
- Bonaccorso E, Peterson AT. A multilocus phylogeny of New World jay genera. Mol Phylogenet Evol. 2007;42:467–476.10.1016/j.ympev.2006.06.025
- Benz BW, Robbins MB, Peterson AT. Evolutionary history of woodpeckers and allies (Aves: Picidae): placing key taxa on the phylogenetic tree. Mol Phylogenet Evol. 2006;40:389–399.10.1016/j.ympev.2006.02.021
- Saunders MA, Edwards SV. Dynamics and phylogenetic implications of MtDNA control region sequences in New World Jays (Aves: Corvidae). J Mol Evol. 2000;51:97–109.10.1007/s002390010070
- Cicero C, Johnson NK. Higher-level phylogeny of New World vireos (Aves: Vireonidae) based on sequences of multiple mitochondrial DNA genes. Mol Phylogen Evol. 2001;20:27–40.10.1006/mpev.2001.0944
- Kennedy JD, Weir JT, Hooper DM, et al. Ecological limits on diversification of the Himalayan core Corvoidea. Evolution. 2012;66:2599–2613.10.1111/evo.2012.66.issue-8
- Jønsson KA, Fabre P-H, Irestedt M. Brains, tools, innovation and biogeography in crows and ravens. BMC Evol Biol. 2012;12:72.
- Feldman CR, Omland KE. Phylogenetics of the common raven complex (Corvus: Corvidae) and the utility of ND4, COI and intron 7 of the beta-fibrinogen gene in avian molecular systematics. Zool Scr. 2005;34:145–156.10.1111/zsc.2005.34.issue-2
- Andersen MJ, Hosner PA, Filardi CE, et al. Phylogeny of the monarch flycatchers reveals extensive paraphyly and novel relationships within a major Australo-Pacific radiation. Mol Phylogen Evol. 2015;83:118–136.10.1016/j.ympev.2014.11.010
- Cracraft J, Feinstein J. What is not a bird of paradise? Molecular and morphological evidence places Macgregoria in the Meliphagidae and the Cnemophilinae near the base of the corvoid tree. Proc R Soc Lond B. 2000;267:233–241.10.1098/rspb.2000.0992
- Kryukov A, Spiridonova L, Nakamura S, et al. Comparative phylogeography of two crow species: jungle crow Corvus macrorhynchos and carrion crow Corvus corone. Zool Sci. 2012;29:484–492.10.2108/zsj.29.484
- Slager DL, Battey C, Bryson RW, et al. A multilocus phylogeny of a major New World avian radiation: the Vireonidae. Mol Phylogen Evol. 2014;80:95–104.10.1016/j.ympev.2014.07.021
- Helm-Bychowski K, Cracraft J. Recovering phylogenetic signal from DNA sequences: Relationships within the corvine assemblage (class Aves) as inferred from complete sequences of the mitochondrial DNA cytochrome-b gene. Mol Biol Evol. 1993;10:1196–1214.
- Bonaccorso E, Peterson AT, Navarro-Sigüenza AG, et al. Molecular systematics and evolution of the Cyanocorax jays. Mol Phylogen Evol. 2010;54:897–909.10.1016/j.ympev.2009.11.014
- Price TD, Hooper DM, Buchanan CD, et al. Niche filling slows the diversification of Himalayan songbirds. Nature. 2014;509:222–225.10.1038/nature13272
- Bonaccorso E. Historical biogeography and speciation in the Neotropical highlands: molecular phylogenetics of the jay genus Cyanolyca. Mol Phylogen Evol. 2009;50:618–632.10.1016/j.ympev.2008.12.012
- Kryukov A, Iwasa M, Kakizawa R, et al. Synchronic east-west divergence in azure-winged magpies (Cyanopica cyanus) and magpies (Pica pica)*. J Zool System Evol Res. 2004;42:342–351.10.1111/jzs.2004.42.issue-4
- Zhang R, Song G, Qu Y, et al. Comparative phylogeography of two widespread magpies: importance of habitat preference and breeding behavior on genetic structure in China. Mol Phylogen Evol. 2012;65:562–572.10.1016/j.ympev.2012.07.011
- Cibois A, Pasquet E. Molecular analysis of the phylogeny of 11 genera of the Corvidae. Ibis. 1999;141:297–306.
- Morinha F, Clemente C, Cabral JA, et al. Next-generation sequencing and comparative analysis of Pyrrhocorax pyrrhocorax and Pyrrhocorax graculus (Passeriformes: Corvidae) mitochondrial genomes. Mitochondrial DNA Part A. 2016;27:2278–2281.
- Gonzalez J, Wink M, Garcia-del-Rey E, et al. Evidence from DNA nucleotide sequences and ISSR profiles indicates paraphyly in subspecies of the Southern Grey Shrike (Lanius meridionalis). J Ornithol. 2008;149:495–506.10.1007/s10336-008-0293-y
- Fuchs J, Crowe TM, Bowie RC. Phylogeography of the fiscal shrike (Lanius collaris): a novel pattern of genetic structure across the arid zones and savannas of Africa. J Biogeogr. 2011;38:2210–2222.10.1111/jbi.2011.38.issue-11
- Primmer C, Borge T, Lindell J, et al. Single-nucleotide polymorphism characterization in species with limited available sequence information: high nucleotide diversity revealed in the avian genome. Mol Ecol. 2002;11:603–612.10.1046/j.0962-1083.2001.01452.x
- Reddy S. Systematics and biogeography of the shrike-babblers (Pteruthius): species limits, molecular phylogenetics, and diversification patterns across southern Asia. Mol Phylogen Evol. 2008;47:54–72.10.1016/j.ympev.2008.01.014
- Sorenson MD, Ast JC, Dimcheff DE, et al. Primers for a PCR-based approach to mitochondrial genome sequencing in birds and other vertebrates. Mol Phylogen Evol. 1999;12:105–114.10.1006/mpev.1998.0602
- Chesser RT. Molecular systematics of the rhinocryptid genus Pteroptochos. Condor. 1999;101:439–446.
- Prychitko TM, Moore WS. The utility of DNA sequences of an intron from the β-fibrinogen gene in phylogenetic analysis of woodpeckers (Aves: Picidae). Mol Phylogen Evol. 1997;8:193–204.10.1006/mpev.1997.0420
- Fjeldsa J, Zuccon D, Irestedt M, et al. Sapayoa aenigma: a New World representative of Old World suboscines. Proc R Soc Lond B. 2003;270:S238–S241.10.1098/rsbl.2003.0075
- Slade R, Moritz C, Heideman A, et al. Rapid assessment of single-copy nuclear DNA variation in diverse species. Mol Ecol. 1993;2:359–373.10.1111/j.1365-294X.1993.tb00029.x
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797.10.1093/nar/gkh340
- Nylander J. MrModeltest v2. Program distributed by the author. Uppsala: Evolutionary Biology Centre, Uppsala University; 2004.
- Gutell RR, Jansen RK. Genetic algorithm approaches for the phylogenetic analysis of large biological sequence datasets under the maximum likelihood criterion; 2006.
- Felsenstein J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution. 1985;39:783–791.
- Paup DS. Phylogenetic analysis using parsimony (* and other methods). Sunderland, MA: Sinauer Associates; 2003.
- Hillis DM, Bull JJ. An empirical test of bootstrapping as a method for assessing confidence in phylogenetic analysis. Syst Biol. 1993;42:182–192.10.1093/sysbio/42.2.182
- Wilcox TP, Zwickl DJ, Heath TA, et al. Phylogenetic relationships of the dwarf boas and a comparison of Bayesian and bootstrap measures of phylogenetic support. Mol Phylogen Evol. 2002;25:361–371.10.1016/S1055-7903(02)00244-0
- Altekar G, Dwarkadas S, Huelsenbeck JP, et al. Parallel metropolis coupled Markov chain Monte Carlo for Bayesian phylogenetic inference. Bioinformatics. 2004;20:407–415.10.1093/bioinformatics/btg427
- Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–1574.10.1093/bioinformatics/btg180
- Ronquist F, Teslenko M, van der Mark P, et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61:539–542.10.1093/sysbio/sys029
- Rambaut A, Suchard M, Xie D, et al. Tracer v1. 6; 2014. Available from: http://beast.bio.ed.ac.uk/Tracer
- Ree RH, Moore BR, Webb CO, et al. A likelihood framework for inferring the evolution of geographic range on phylogenetic trees. Evolution. 2005;59:2299–2311.10.1111/evo.2005.59.issue-11
- Ree RH, Smith SA. Maximum likelihood inference of geographic range evolution by dispersal, local extinction, and cladogenesis. Syst Biol. 2008;57:4–14.10.1080/10635150701883881
- Yu Y, Harris A, He X. S-DIVA (Statistical Dispersal-Vicariance Analysis): a tool for inferring biogeographic histories. Mol Phylogen Evol. 2010;56:848–850.10.1016/j.ympev.2010.04.011
- Yu Y, Harris AJ, Blair C, et al. RASP (Reconstruct Ancestral State in Phylogenies): a tool for historical biogeography. Mol Phylogen Evol. 2015;87:46–49.10.1016/j.ympev.2015.03.008
- Ronquist F. Dispersal-vicariance analysis: a new approach to the quantification of historical biogeography. Syst Biol. 1997;46:195–203.10.1093/sysbio/46.1.195
- Nylander JA, Olsson U, Alström P, et al. Accounting for phylogenetic uncertainty in biogeography: A Bayesian approach to dispersal-vicariance analysis of the thrushes (Aves: Turdus). Syst Biol. 2008;57:257–268.10.1080/10635150802044003
- Smith SA. Lagrange C++ Manual; 2010.
- Ronquist F. Bayesian inference of character evolution. Trends Ecol Evol. 2004;19:475–481.10.1016/j.tree.2004.07.002
- eBird. eBird: an online database of bird distribution and abundance. eBird; 2015. Available from: http://www.ebird.org
- Drummond AJ, Suchard MA, Xie D, et al. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 2012;29:1969–1973.10.1093/molbev/mss075
- Parham JF, Donoghue PC, Bell CJ, et al. Best practices for justifying fossil calibrations. Syst Biol. 2011;61:346–359.
- Hosner PA, Nyári ÁS, Moyle RG. Water barriers and intra-island isolation contribute to diversification in the insular Aethopyga sunbirds (Aves: Nectariniidae). J Biogeogr. 2013;40:1094–1106.10.1111/jbi.12074
- Andersen MJ, Shult HT, Cibois A, et al. Rapid diversification and secondary sympatry in Australo-Pacific kingfishers (Aves: Alcedinidae: Todiramphus). R Soc Open Sci. 2015;2:140375.10.1098/rsos.140375
- Lerner HR, Meyer M, James HF, et al. Multilocus resolution of phylogeny and timescale in the extant adaptive radiation of Hawaiian honeycreepers. Curr Biol. 2011;21:1838–1844.10.1016/j.cub.2011.09.039
- Fox DL. Growth increments in Gomphotherium tusks and implications for late Miocene climate change in North America. Palaeogeogr Palaeoclimatol Palaeoecol. 2000;156:327–348.10.1016/S0031-0182(99)00148-0
- Kohn MJ, Fremd TJ. Miocene tectonics and climate forcing of biodiversity, western United States. Geology. 2008;36:783–786.10.1130/G24928A.1
- Sanmartín I, Enghoff H, Ronquist F. Patterns of animal dispersal, vicariance and diversification in the Holarctic. Biol J Linnean Soc. 2001;73:345–390.10.1111/j.1095-8312.2001.tb01368.x
- Moyle RG, Oliveros CH, Andersen MJ, et al. Tectonic collision and uplift of Wallacea triggered the global songbird radiation. Nat Comm. 2016;7:12709.10.1038/ncomms12709
- ODea A, Lessios HA, Coates AG, et al. Formation of the Isthmus of Panama. Sci Adv. 2016;2:e1600883.10.1126/sciadv.1600883
Appendix 1. List of samples used in the study.
Notes: GenBank sequences from previous studies are denoted by ‘G’. Institutional abbreviations: AMNH, American Museum of Natural History; KUNHM, University of Kansas Natural History Museum; LSUMNS, Louisiana State University Museum of Natural Science; NTNU, National Taiwan Normal University.
Appendix 2.
Figure 2A. Phylogeny based on Bayesian and maximum-likelihood analyses of the combined nuclear data set. Numbers at the nodes refer to Bayesian posterior probability/maximum-likelihood bootstrap support.
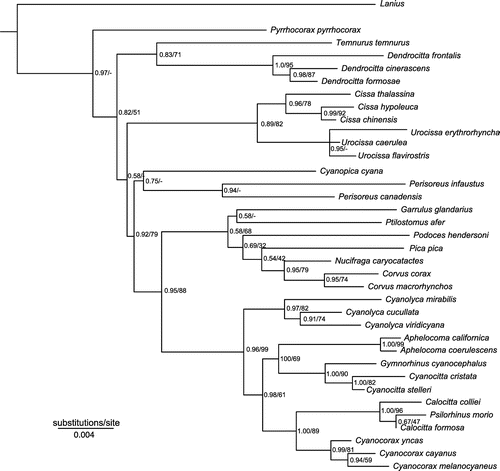
Figure 2B. Phylogeny based on Bayesian and maximum-likelihood analyses of the combined mitochondrial data set. Numbers at the nodes refer to Bayesian posterior probability/maximum-likelihood bootstrap support.