
ABSTRACT
Laboratories that perform PCR on a routine basis need to count on reliable DNA isolation methods. In locations where supply of DNA extraction kits depends on importation, having an in-house protocol is desirable. This is also important for laboratories limited by budget constraints. We present a low-cost DNA isolation protocol that incorporates well-known techniques, but that we have adapted to various animal tissues. We tested this protocol on animal blood and muscle, and on cell suspension from skin swabs. The results were comparable, in terms of amount and quality of DNA, to those obtained with two other commercially available methods. DNA retrieved with this protocol has been successfully employed for Sanger sequencing of gene PCR products from animal tissues and blood, as well as for PCR-based diagnosis of chytrid fungus in amphibians and blood parasites in birds.
Introduction
Every laboratory that conducts DNA isolation on a regular basis seeks to count on standardized protocols tailored to its needs. It is desirable that such a protocol, besides being effective in terms of quantity and quality of the isolated genomic material, is also cost-effective. This is especially relevant in countries where commercial kit availability depends on importation, which implies higher costs and might result in a forced delay until reagent arrival.
Here, we describe a cost-effective protocol that we have used routinely for DNA isolation from diverse animal tissues. The present protocol resembles commonly used protocols (e.g [Citation1].) in that it brings together well-known techniques for DNA isolation, such as initial tissue degradation with the aid of proteinase K in presence of SDS and EDTA [Citation2], protein denaturation by a guanidine salt [Citation3], and final DNA precipitation with isopropanol. Nevertheless, to our knowledge, no other published protocol combines solutions and reagents in the way that we present here, hence our intent to share it with other low-budget laboratories that may benefit from it. Additionally, we have made several modifications to the original protocol and have extended its applicability to other types of samples besides regular animal tissue (i.e. liver and muscle), and provide troubleshooting tips to deal with different experimental situations. Using this protocol, we have successfully retrieved DNA from muscle, skin, and liver of vertebrates for downstream PCR applications in phylogenetics [Citation4], phylogeography [Citation5], and diversification [Citation6], but we have also adapted the protocol to isolate DNA from avian blood samples kept in 99% ethanol for malaria detection [Citation7] and from skin swabs of amphibians to detect Batrachochytrium dendrobatidis (Bd) infection [Citation8].
Methods
In order to show the versatility and effectiveness of our protocol, we extracted genomic material from muscle tissue (silky shark [Carcharhinus falciformis]; tissues LBE-192, 218, and 271), avian blood (eared dove [Zenaida auriculata]; tissues HFC-S-1072, 1529, and 1532), and amphibian skin swabs (Cane toad [Rhinella marina]; tissues LC-001–003). All samples belong to the tissue collection held at Laboratorio de Biología Evolutiva, Universidad San Francisco, Quito, Ecuador. We evaluated the performance of our protocol in comparison to commercially available DNA isolation kits that either use (DNeasy Blood and Tissue Kit by QIAGEN) or do not use (Wizard Genomic DNA Purification Kit by Promega) membrane columns in their protocols.
Muscle and blood samples were preserved in 96% ethanol at −20°C, whereas swabs were stored in dry Eppendorf vials at −20°C. Muscle samples were cut in three pieces of approximately the same size and wet weight of each piece was recorded (). In the case of blood samples, blood was resuspended and homogenized in the alcohol solution, prior to splitting in three equal volumes. All subsamples were then centrifuged, supernatant alcohol was discarded, and the blood pellet was allowed to dry before its weight was recorded (this follows the initial steps of our protocol; see Protocol section). For the sake of comparing our protocol to commercial kits, swab samples had to be treated differently than indicated in our protocol. Each swab was immersed in 900 µL of nuclease-free water (Invitrogen) and mixed by vortex to promote the cells collected by the swab to come into suspension. Then, an aliquot of 300 µL of cell suspension was the starting material for each extraction method. Normally, this step is not required, since the swab comes into contact directly with the lysis solution (see Protocol section).
Table 1. Comparison of this protocol to two commercially available DNA isolation kits in terms of amount and quality of DNA that is extracted from three different tissues of three animal species.
We followed the instructions of the commercial kits as specified by the manufacturers, applying the corresponding methods for blood and tissue DNA extraction. In the case of the swab cell suspension, the sections “Animal Tissue (Spin-Column Protocol)” and “Isolating Genomic DNA from Tissue Culture Cells and Animal Tissue” from the DNeasy kit and the Wizard kit, respectively, were used. In order to visually verify the presence of DNA material, 3 µL of the final elution was loaded in a 1.0% agarose gel for electrophoresis. DNA yield and quality were measured with a Biotek EPOCH Microplate Spectrophotometer apparatus.
Results and discussion
DNA yield
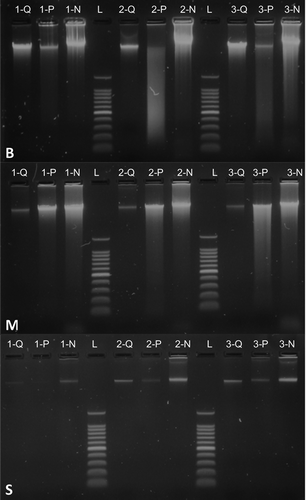
The output of the protocol presented here is comparable to that of commercially available DNA isolation kits. For each tissue type, and for every sample, our method was able to retrieve enough genomic material as to detect it through agarose electrophoresis (). Visual inspection via electrophoresis suggested that our protocol was able to isolate more genomic material every time, which was confirmed by spectrophotometric measurements (the only exception being sample LC-001) (). As expected, the amount of DNA obtained from swabs was considerably lower than from the other tissues due to the nature of the sampling method, which collects loose cells from the skin of amphibians.
Figure 1. Gel electrophoresis of DNA isolated from three animal tissue types by three different protocols. Top down, the panels show DNA extractions from avian blood (B), shark muscle (M) and amphibian skin swabs (S). Numbers correspond to different individuals of eared dove (Zenaida auriculata) (panel B), silky shark (Carcharhinus falciformis) (panel M), and cane toad (Rhinella marina) (panel S). Letters correspond to the three methods employed: DNeasy Blood and Tissue Kit by QIAGEN (Q), Wizard Genomic DNA Purification Kit by Promega (P) and our protocol (N). The molecular-weight size marker (L) used was the 100 bp DNA Ladder by Promega, the heaviest fragment of which is 1500 bp.
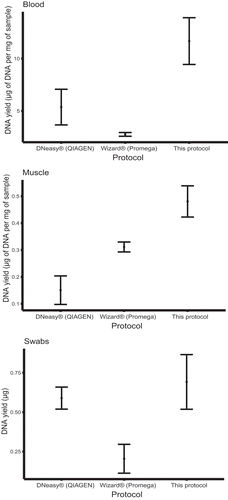
In order to test the performance of our protocol in comparison to the two commercial kits, we determined the ratio of amount of DNA extracted to the amount of initial sample. Here, we took into account blood and muscle samples only, since we were not able to determine the approximate number of cells in suspension from swab samples. Our protocol surpassed the other two methods in terms of µg of isolated DNA per mg of sample (for blood and muscle tissue). The same holds true in the case of total µg of DNA extracted from swab samples ().
Figure 2. Amount of total DNA (µg) extracted from different animal tissues with three different protocols. For blood and muscle samples, amount of DNA extracted, relative to the initial amount of sample is presented.
DNA quality
Electrophoresis images showed that the DNA extracted with our protocol was not as clean as that extracted from commercial kits, especially as the one extracted with DNeasy protocol (). This result was expected given the use of a silica-based membrane, which promotes DNA adsorption only [Citation9]. We assessed the quality of the extracted DNA in terms of absorbance ratios at 230 nm, 260 nm and 280 nm (). The ratio of absorbance at various wavelengths is typically used as an indicator of purity of nucleic acid solutions. The ratio of 260 nm over 280 nm expresses the quality of DNA and RNA. A ratio of ~1.8 and of ~2.0 is accepted to represent good quality for DNA and RNA, respectively. Values considerably lower than 1.8 are commonly associated with the presence of contaminants (e.g. proteins), which have high absorbance around 280 nm. Absorbance ratio at 260 nm/230 nm is usually used as a secondary criterion for nucleic acid purity. A value between 2.0 and 2.2 is regarded as representative of good quality, lower values indicating typically contamination with other compounds (probably related to the extraction procedure). We expected our protocol to achieve lower quality scores because it has not undergone rigorous examination in order to comply with quality standards that are compulsory for commercial products. Nevertheless, our protocol fared reasonably well in terms of the quality of the DNA extracted ().
Table 2. Comparison between this protocol and two commercial DNA extraction kits in terms of average quality of isolated DNA from three different tissues. Quality was assessed by calculating ratios of absorbance measured at 230 nm, 260 nm and 280 nm. A referential scale is also given: ++ optimal or near optimal quality; +- suboptimal quality; – poor quality (relative to ranges discussed in the text).
We are aware that the size of our statistical sample is extremely reduced (n = 3), and therefore caution has to be exercised when drawing conclusions. Nevertheless, our goal was never to assess superiority in performance among the three methods evaluated here, but to demonstrate that our protocol is able to retrieve quantities of genomic material comparable to those extracted with popular isolation kits in the market. We have shown evidence on the efficiency of this protocol in effectively extracting genetic material form different animal sources. In quantity and quality of the isolated DNA, this protocol rivals the output of existing commercial methods at a fraction of their cost, thus making it especially appealing to low-budget laboratories.
Protocol
Note: We have highlighted in bold our contributions and/or modifications to the original protocol.
Reagents and solutions
The protocol for DNA extraction uses the following reagents and solutions: sodium chloride (NaCl), ethylenediaminetetraacetic acid disodium salt dihydrate (EDTA), tris(hydroxymethyl)aminomethane (Tris), sodium dodecyl sulfate (SDS), guanidine isothiocyanate, Proteinase K solution (20 mg/mL), 70% ethanol solution, 100% isopropanol, Lysis Solution (NaCl 1M, Tris 100 mM pH 8.0, EDTA 25 mM pH 8.0, SDS 0.5%; sterilized by autoclave), Protein Precipitation Solution (guanidine isothiocyanate 4M and Tris 0.1M pH 7.5; sterilized by filtration), and RNase A solution (20 mg/mL). When filter-sterilizing the Protein Precipitation Solution does not use cellulose acetate filters, since they will dissolve. Use 0.22 µm cellulose nitrate, polyethersulfone or mixed cellulose esters membranes instead. For troubleshooting (see ) the protocol may need 4M sodium acetate solution.
Table 3. Troubleshooting guide for the present DNA extraction protocol.
Procedure
In order to dissolve any crystals that may have precipitated in the Lysis Solution during storage, it is advisable to heat it before use. Typically, 1–2 min microwave heating (medium power) will be enough. Proceed according to the following steps:
1.Digest biological sample in Lysis Solution
a. Muscle/Liver:
i. Cut into small pieces 5–20 mg (wet weight) and place them inside a 1.5 mL Eppendorf vial containing 300 µL of the Lysis Solution.
ii. Grind the pieces inside the tube with a small mortar pestle.
b. Blood (preserved in 99% ethanol):
i. Transfer 1 mL of the blood-ethanol mix to a 1.5 mL Eppendorf vial.
ii. Centrifuge the vial at ~16,000 g (or at 13,000 rpm in an 8 cm-radius centrifuge) for 2 min to bring all blood particles to form a pellet.
iii. Make sure to remove all remaining ethanol with a micropipette and allow to airdry by leaving the vial’s lid open for 5 min.
iv. Pour 300 µL of the Lysis Solution into the vial.
c. Swabs:
i. Cut the tip of the swab and place it inside a 1.5 mL Eppendorf vial containing 400 µL of the Lysis Solution.
ii. Vortex vigorously for 2 min.
iii. Take out the swab. The swab can be kept at −20 ºC for future extraction attempts.
2. Add 3 µL of Proteinase K Solution and mix by vortexing (when working on Batrachochytrium dendrobatidis detection, use 15 µL of Proteinase K Solution, instead).
3. Incubate the sample in a thermo shaker at 55 ºC and 900 rpm*; 15–18 h (overnight) for tissue and blood; 2–3 h for swab samples, or for small blood or tissue samples (~5 mg) making sure that the whole sample is completely digested.
*It is highly recommended to digest the samples using a thermo shaker with a speed of 900 rpm, or above, to maintain the particles in solution and prevent them from depositing at the bottom of the tube (which delays tissue digestion significantly). If a thermo shaker is not available, regular mixing by vortexing (every 20–30 min) is required. If the samples are left overnight, add 3 µL of Proteinase K solution and mix by vortexing, before incubating.
4. After incubation at 55 ºC, heat the vial at 95 ºC for 10 min to inactive any residual Proteinase K.
5. Allow the samples to cool at room temperature for 5–10 min and add 4 µL of RNase A solution to the lysate and mix by tipping the bottom of the tube firmly and repeatedly.
6. Incubate the samples at 37 ºC for 30 min.
7. Leave the tubes to cool at room temperature for 5 min.
8. Add 100 µL of Protein Precipitation Solution and vortex vigorously for 20 s.
9. Centrifuge the tubes at ~16,000 g for 10 min.
10. Carefully transfer the supernatant by pouring it into another empty 1.5 mL tube without disturbing the residual pellet.
It is important to avoid transferring any particle of the pellet along with the supernatant. In that case, another shorter (5 min) centrifugation step is needed, followed by further recovering of remaining supernatant.
11. Precipitate the DNA by adding 300 µL of chilled 100% isopropanol (kept inside the −20 ºC freezer). Mix well by flipping the tubes several times, until the DNA pellet is visible.
12. Centrifuge at ~16,000 g for 2–5 min.
If a large amount of DNA is easily noticeable in the pellet after a short centrifugation (i.e. 2 min), a longer centrifugation time is not necessary. On the contrary, if DNA is not visible yet or a small amount of it is expected, keep centrifuging for up to 10 min.
13. Discard the supernatant and add 300 µL of 70% ethanol. Wash the DNA pellet by flipping the tubes 5–10 times.
14. Centrifuge at ~16,000 g for 2–5 min.
Before proceeding with centrifugation make sure that the DNA pellet is not attached to the tube walls or cap.
15. Discard the supernatant by absorbing with a micropipette (better with filter tip) and leave the DNA pellet as dry as possible.
16. Leave air-dry the tubes open until no drops of liquid are visible.
17. Resuspend the DNA pellet in any preferred rehydration solution (i.e. TE 0.1X pH 8.0 or Tris-Cl 10 mM pH 8.0).
18. Incubate at 65 ºC for 1 h. Occasional tapping the bottom of the vial is recommended.
19. Leave the tubes at 4 ºC overnight for complete resuspension.
At this point the vial containing the DNA solution can be preserved at −20 ºC for long-time storage or kept at 4 ºC for subsequent PCR. Troubleshooting tips are available in .
Acknowledgments
To Charles Linkem for sharing a basic DNA extraction procedure, initially developed by M. Fujita. The work of Gabriela Gavilanes, Gabriela Nichols, María Eugenia Ordoñez, Nathalia Valencia, and several graduate and undergraduate students have helped adapting the protocol to our needs. Research permits and access to genetic resources were granted by Ministerio del Ambiente, Ecuador (MAE-DNB-CM-2016-0041-M-003, No 15-2013-RIG-FAU-DPAP-MA and MAE-DNB-CM-2018-0105).
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- MacManes M. MacManes salt extraction protocol. Figshare J contrib. 2013. doi:10.6084/m9.figshare.658946.v1.
- Gross-Bellard M, Oudet P, Chambon P. Isolation of high-molecular-weight DNA from mammalian cells. Eur J Biochem. 1973;36(1):32–38.
- Bowtell DD. Rapid isolation of eukaryotic DNA. Anal Biochem. 1987;162:463–465.
- Arteaga A, Mebert K, Valencia JH, et al. Molecular phylogeny of Atractus (Serpentes, Dipsadidae), with emphasis on Ecuadorian species and the description of three new taxa. ZooKeys. 2017;661:91–123.
- Prieto-Torres DA, Cuervo AM, Bonaccorso E. On geographic barriers and Pleistocene glaciations: tracing the diversification of the Russet-crowned Warbler (Myiothlypis coronata) along the Andes. PLoS ONE. 2018;13:e0191598.
- Guayasamin JM, Hutter CR, Tapia EE, et al. Diversification of the rainfrog Pristimantis ornatissimus in the lowlands and andean foothills of Ecuador. PLoS ONE. 2017;12(3):e0172615.
- de Aguilar JR, Castillo F, Moreno A, et al. Patterns of avian malaria infection vary with time, but not habitat, in a fragmented Neotropical landscape. PLoS ONE. 2018;31(10):e0206493.
- Tapia EE, Coloma LA, Pazmiño-Otamendi G, et al. Rediscovery of the nearly extinct longnose harlequin frog Atelopus longirostris (Bufonidae) in Junín, Imbabura, Ecuador. Neotrop Biodivers. 2017;3(1):157–167. .
- QIAGEN. DNeasy tissue and blood handbook. Qiagen Group;2006.