
ABSTRACT
Biotechnology is gaining priority along with other rapidly evolving disciplines in science and engineering due to its potential for innovating the modern military. The broad nature of biotechnology is directly relevant to the military and defence sector where the applications span clinical diagnostics, medical countermeasures and therapeutics, to environmental remediation and biofuels for energy. Although the process for a commercial biotech research and development (R&D) pipeline and the Department of Defence (DOD) acquisition cycle both aim to result in products, they follow two distinctly different pathways. In the biotech industry, the pipeline progresses from basic to applied science that includes design and R&D, commercialisation and product launch, where market forces and financial returns on investment drive priorities. Along the way, the scientific and iterative nature of R&D often results in several candidates for a given assay, drug, therapeutic or vaccine, many of which are unsuccessful or wind up in the so-called valley of death. The DOD acquisition process is a multi-phase and often multi-decade cradle-to-grave product lifecycle engrained in mission requirements, warfighter needs and creating legacy programmes of record. The biotech industry is composed of many small R&D and ‘big pharma’ companies that meet DOD’s unique medical mission requirements. These small R&D companies considered that non-traditional DOD acquisition partners are developing new innovations in biotechnology, but the complex DOD acquisition process is challenging for these small start-ups to navigate. Technology solutions that gain support through DOD acquisitions are able to successfully develop their products and bridge the valley of death by obtaining much needed funding for advanced development, test and evaluation, and demonstration through clinical trials. Our analysis profiles three case histories involving private-public partnerships that yielded biotech products developed through the DOD acquisition cycle that continues to meet current and future medical mission requirements.
Introduction-biotechnology does not fit in traditional defence acquisition
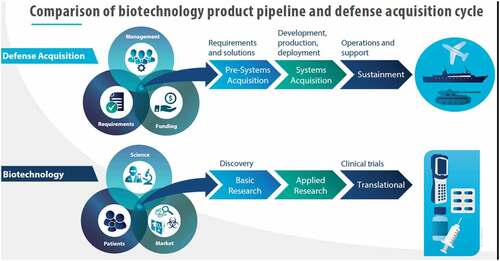
The field of biotechnology is broad and complex especially with medical applications encompassing diagnostics, therapeutics and vaccines which are heavily regulated for efficacy and safety. As a result, industry must prioritise research and development (R&D) investments and balance risk-reward outcomes to increase the chance for successful products in a typical R&D pipeline (). This is evident with countless biotech companies forming through various combinations of technical expertise, strong leadership and great ideas, with the successful ones demonstrating skill in business and financial strategy (Tsai et al., Citation2006). With more biotech products gaining United States (US) Food and Drug Administration (FDA) regulatory approval, higher sales have resulted, while the diversity in partnerships and investments offers greater potential for product development (Evens, Citation2016). The major contrast between the biotech and traditional defence industry is that science and market drives biotech, whereas mission and budget drive defence (Du, Citation2020). In addition, the science of biotechnology deals with living things which will continue to expand the field in areas such as synthetic biology.
Figure 1. Biotechnology product pipeline vs. the defence acquisition cycle. Products are the result of different approaches where biotechnology emphasises discovery and defence emphasises requirements.
The US Department of Defense (DOD) conducts research, development, testing and evaluation (RDT&E) in support of its mission requirements and receives approximately 40% of the total US Government (USG) R&D funds (Sargent Citation2020). DOD and the Department of Health and Human Services (HHS) are the two primary recipients of USG RDT&E funding with DOD normally receiving a higher share such as $600 M in civilian biodefense applications than HHS annually (Watson et al., Citation2018). COVID-19 was the exception with HHS receiving more funding due to the enormous civilian homeland application. HHS also prioritised funding to expedite COVID-19 testing through BARDA and NIH partnership for RADx (Tromberg et al., Citation2020). Historically, the USG tends to fund more basic and applied research where industry tends to fund development that advances product maturity.
The COVID-19 pandemic exemplifies that the bioeconomy in turn is essential and a major driver of our overall economy and reinforces the importance of having a resilient biotech and life sciences industry (NAS, Citation2020; and Weaver, Citation2018). To ensure and sustain viability, aspects of the DOD acquisition process need to be better tailored for biotech especially where bioeconomy industry outputs and DOD acquisition process are not aligned (Du, Citation2020, DiEuliis et al., Citation2020). While the so-called acquisition reform would be difficult, the DOD process would benefit from biotech’s science-based applications and firsthand innovative knowhow, which are hard to implement in a myriad of existing doctrine, oversight and redundancy (Peeler, Citation2016). In addition, the Federal Acquisition Regulation (FAR), which is based on the Code of Federal Regulations, is a traditional framework and basis for governing USG contracting As a procurement mechanism, the FAR has a complex process for meeting statutory mandates, cost accounting compliance, and gate-stages, all of which are not well suited to funding biotech R&D. The speed and innovation required to create value for US businesses needs to be balanced with USG requirements for an effective bioeconomic response.
The typical DOD acquisition model, conventionally used for major, multi-year programs such as aircraft, ships and weapons systems, as well as for major medical countermeasures, is multi-phased and multi-step to include development of and adherence to requirements, achievement of milestones and deliverables, and successful progress reviews. Joint service stakeholders are involved in all phases to help provide critical voice of customer input as a product or technology matures through its project aims and goals. The whole DOD acquisition system is actually composed of three intertwined processes: requirements, funding and actual acquisition (). Ideally, a strategy that hybridises a biotech pipeline and DOD acquisition would lead to greater efficiencies.
Case histories demonstrating successful biotech products via US DOD acquisition
Here, we examine three case histories which included two related to medical treatments for viral diseases and one related to detection and identification of biological warfare agent (BWA) and other infectious disease threats. Our discussion narrates the dynamic between commercial companies interacted and the US Government to make these products available to the market.
Case History 1: Development of Gilead’s Remdesivir through public-private partnership
Remdesivir, which is sold under the brand name Veklury, is an antiviral medication developed by Gilead Sciences, Foster City, CA, to treat COVID-19 in patients who are at least 12 years of age and 88 pounds in weight (39.9 kilograms). Prior to Remdesivir’s repurposing for COVID-19 indication, the programme initially started as a potential treatment for Hepatitis C. In the 2000s, Gilead began basic research on remdesivir’s parent compounds without USG funding and successfully identified the lead candidate in 2013 during its drug discovery process. The US CDC and DOD’s USAMRIID also initiated preclinical studies with Gilead in 2013 and 2014, respectively (Government Accountability Office, Citation2021). The USG later signed an agreement to investigate its therapeutic use in RNA-based filoviruses family for Ebola and coronaviruses family for MERS and SARS. Gilead provided blinded compounds to CDC and USAMRIID, which in turn tested the compounds for antiviral activities against various viruses in their own highly specialised laboratories. Similarly, in 2014, the US NIH provided ed funding to Gilead, which supplemented their existing R&D for further investigation on remdesivir’s use on coronavirus
While this public-private partnership was successful because remdesivir turned out to be an effective treatment during the COVID-19 pandemic, the collaboration relied on earlier investments that could be repurposed with minimal USG intervention which offered Gilead flexibility to reinforce its own scientific innovation and best practices. Among the total $162 million USG funding for remdesivir, about $40 million came from DoD, $121 million from NIH and $0.7 million from the CDC (Government Accountability Office, Citation2021). At the same time, Gilead invested about $1.3 billion as of December 2020.
In 2016, Gilead entered into an Other Transaction Authority (OTA) contract vehicle as part of a prototyping agreement with DOD’s Joint Project Manager for Medical Countermeasure Systems (JPM-MCS) to study remdesivir’s efficacy for treating Ebola and other filoviruses. The award potential is upwards to roughly $50 million to support animal testing and procuring a certain number of doses for national stockpiling, and as of December 2020, $33 million has been obligated.
Case History 2. Joint Biological Agent Identification and Diagnostic System (JBAIDS)
Rapid diagnosis of biological threat agents whether from an accidental, intentional or naturally occurring event has long been a challenge in the battlefield or hospital setting. Regarding clinical diagnosis, there is a continual need for clinical laboratory-quality result and identification of biological agent with a sample to answer turnaround time of hours rather than days typical of traditional culture methods. The Joint Biological Agent Identification and Diagnostic System (JBAIDS) entered its System Development stage in 2003 and became a landmark for facilitating biotech product development through the DOD acquisition process. JBAIDS, which is now a DOD program of record, fielded the first ever FDA-cleared system for anthrax across the US Armed Forces for the Air Force, Army, Marine Corps and Navy.
The JBAIDS system was the result of a five-year, $100 M+ contract award to Idaho Technology, Inc. (Idaho Technology) of Salt Lake City, UT. In 2003, the Joint Program Executive Office for Chemical Biological Defense (JPEO CBD) established a spiral-development acquisition program of record for a field-deployable biowarfare agent diagnostic system. The Block 1 iteration involved the revision of an existing commercially available instrument into a field-deployable laboratory diagnostic system with initial instrument hardware and several molecular-based diagnostic assay kits that can be simultaneously run to detect various BWA. JBAIDS uses quantitative polymerase chain reaction (qPCR), which was qualified as the core technology and identifies specific BWA according to the presence of that target’s nucleic acid. Block 1 requirements included identifying up to 10 different target agents simultaneously in under 40 minutes, which is a typical run time for qPCR. Detection and identification of various BWA were demonstrated across multiple relevant clinical samples such as blood, nasal swabs, sputum and urine (Wilson, Citation2006). DOD later replaced the Block 2, 3 iterations when they changed their programmatic priorities.
JBAIDS demonstrated that the DOD acquisition model, known as the DOD 5000 Series, conventionally used to develop and acquire major weapon systems, vehicles and other military materiel, is flexible and can be tailored for unique medical products that require FDA regulatory oversight. The DOD acquisition process is a phased and sequential set of events used to manage defence programs that fulfill the needs of the DOD. The JBAIDS program office developed its acquisition strategy and plan according to DOD 5000.2 Source Selection guidelines. This involved sourcing and selecting a qualified contractor, planning and executing a development and testing phase to verify the system met all requirements, conducting operational testing to ensure it met the user needs (Wilson, Citation2006). In addition, the project plan had to include regulatory pathway elements to verify and validate the product in accordance with FDA guidance in order to obtain FDA clearance. For expediency, the JBAIDS development and testing for hardware and assays were executed in parallel, which cut the acquisition time in half, enabling production and fielding with FDA clearance in an unusually short timeline.
Earlier, Idaho Technology had developed and validated their core pathogen detection technology through self-funded efforts and SBIR grants focusing on clinical and molecular diagnostics for the Air Force under DOD funding and driven by commercial market forces. Using what is known as a modified commercial-off-the-shelf acquisition, the DOD leveraged these prior investments to achieve its needs and meet its technical objective for a common diagnostic platform with dual use in commercial applications as well as unique DOD needs using modern molecular methods (Wilson, Citation2006). The initial idea began at Idaho Technology with an approach of taking a proven laboratory-grade PCR instrument to a battlefield or military operational setting. However, there were significant challenges for making a portable, rugged instrument with ready-to-use reagent kits that did not require cold-chain storage and providing standard operating procedures for field use with basic operator interfaces. Working closely with the Air Force in the early years, this collaboration produced a field laboratory PCR device called the RAPID and gave Idaho Technology the familiarity with DOD RDT&E and contracting which later became valuable for forming its partnership with the JBAIDS program.
The JBAIDS Source Selection had four phases that began after first conducting a market survey to determine the most feasible technology that existed according to the JRO’s Common Diagnostic System requirements. In order to meet the requirements for high sensitivity (test accuracy) for detecting and correctly identifying an agent and fast, sample-to-answer turnaround time, PCR was chosen as the Block 1 technology of choice (Wilson, Citation2006). The Source Selection process involved an initial down-selection of industry bidders who bid suitable technology according to the published requirements, and then final potential candidates were notified and invited to participate in a competitive testing or ‘fly off’. Finally, a request for proposal (RFP) was issued to those successful bidders in the fly-off. The overall source selection required a significant investment by each bidder to participate in the fly-off. The competitive fly-off consisted of a two-week event held at the US Army Dugway Proving Ground in Utah where select agents could be provided and properly handled at their facilities.
Idaho Technology was the ultimate selectee from this down-selection, and upon contract award, they began the advanced development and demonstration of their system to meet DOD requirements, participated in functional and operational assessments and prepared and submitted a data package to the FDA for 510(k) clearance of the anthrax in vitro diagnostic kit. Upon successful completion of these milestones, Idaho Technology began initial low-rate and then full-rate production for delivery to the four services according to their mission needs.
As a DOD program of record, JBAIDS became the legacy diagnostic system for 15 years, meeting the need for a common, standardised biothreat agent diagnostic system across the US Armed Forces, until its replacement − the Next-Generation Diagnostic System (NGDS) − was fielded. The NGDS was designed to be more portable and field-deployable with a smaller footprint and simpler workflow evolving from the initial JBAIDS increment − now retired.
Case history 3. Medical Countermeasures for Viral Haemorrhagic Fevers (VHF)
Often, the USG must support infectious disease threat areas where the commercial market is not available or investments in basic and applied research are limited to fully develop medical countermeasures. An example has been the 40+ year investments focused on viral haemorrhagic fevers (VHF), specifically Ebola virus disease. The first cases were identified in 1976, and soon afterwards, the DOD invested in research to better understand the virus’ lethality and develop MCM for detection, prevention and treatment. In addition, maximum Biosafety Level-4 high biological containment, which is expensive, laborious and meticulously performed with other government laboratories, was a requirement. Over the course of this effort, a better understanding of the virus and other filoviruses was developed which included animal models of disease that are essential to assess MCMs being developed. Consequently, the evaluation of vaccines, therapies and assays to detect the virus and host responses were developed. Unfortunately, many traditional approaches such as inactivated virus vaccines and some early subunit vaccines were not effective in protecting against this virus infection. In the last 15 years, DOD funded a special program called the Transformational Medical Technologies Initiative (TMTI) through the Defense Threat Reduction Agency as well as HHS, which led to a variety of vaccine candidates and therapeutic approaches. Public private partnerships were essential for the advancement of these MCMs and were often small biotech companies with emerging technological approaches. These efforts included the development and validation of molecular real-time PCR methods developed for use during a potential Emergency Use (Whitehouse et al., Citation2015). Essentially, these assays were pre-positioned using animal samples and the relatively few human samples that were collected during outbreaks. Unfortunately, despite success with various vaccine platforms and therapeutic approaches, there was limited funding available from DOD or DHHS to complete IND enabling studies, GMP production and phase I clinical safety testing. Often, the funding was not concurrent with the development stage of the drug and the funding was available before the approach was maximised. While developers and funders were aware of the limited funding, the goal was to place the MCM in a position so it could be used in an outbreak or pandemic situation as was done for the molecular diagnostic assay. Alternative approaches with some commercial value could have been used such as traveller’s vaccination and ecological use for protecting apes and chimpanzees. For one candidate called ZMAPP, the cost for GMP production, pre-clinical testing and a small phase I clinical testing was less than $1 million USD. Following BSL-4 laboratory accidents, the VSV vaccine candidate was used as a prophylaxis therapy, but the safety and efficacy were inconclusive due to the limited numbers involved. Hence, the investment would fall short of being placed on EUA readiness stage of development. The products would therefore languish in the purgatory waiting for the eventual product death as opposed to being shelved and ready for use if needed. During the 2014–2015 Ebolavirus Disease in West Africa, the molecular assay would be part of the U.S. contribution for early diagnostic assays in West Africa and infected citizens returning to the US. ZMAPP was the first treatment utilised in an infected US citizen and became a promising treatment co-developed by the U.S. and Canadian government that otherwise languished in the valley of death for about four years. In retrospect, it was clear that pre-clinical data on the product and phase I human clinical studies demonstrating safety would have made the decision to treat patients less controversial.
Lessons learned
Despite all the successful R&D and commercialisation efforts as demonstrated in our case histories, DOD seldom requests a procurement contract despite all the approvals, qualifications and relationships. As a result, industry often interprets a mixed message that the USG is interested in procuring the products but has no idea when and how. This frustrates many startups that lack cash reserve. Thus, DOD’s acquisition process is unable to send a clear market demand signal to help companies and investors make better business projections. The negative impacts could be products with multi-use applications not being fully leveraged, and even worse promising talents and innovations are forced out of business or acquired at an unfair value by buyers with adversarial intent. This trend is unlikely to change soon with the rate of technology continuing to accelerate and startups with limited resources and capacity for R&D and ensuing product development. Less than 40% of contractor independent R&D is applied to DOD Modernisation priorities for artificial intelligence/machine learning, autonomy, biotechnology, cyber, directed energy, fully networked command, control, and communications, hypersonics, microelectronics, quantum science and space (Government Accounting Office, Citation2020). Further communication is needed to better align contractor independent R&D with DOD Modernisation priorities.
As mentioned, the FAR is often used for large contracting where it serves as rule of law for administering procurements. Other Transaction Authority (OTA) contracts have become frequent alternatives because they are more agile funding vehicles than traditional FAR-based contracts. (Smith & Diggans, Citation2020). OTAs, which are often used for R&D and direct access to commercial products, are more flexible contract vehicles making them convenient to expedite mission critical elements (Ardizzone & Love, Citation2020; McCormick, Citation2020). The partnership between Gilead and DoD laboratories was synergistic because the OTA and related cost share could be further leveraged and pivoted to other potential targets, including coronaviruses family. One innovative feature in the OTA is that the agreement stipulating it is not subject to the Bayh-Dole Act, a USG legislation that may allow the USG certain rights on inventions arising from federally funded research. For example, the remdesivir findings covered in this agreement are not considered subject invention by the government, and the USG will not be granted March-In Rights to force licence remdesivir to other companies. In addition, DOD, NIH and academic researchers concluded that the USG support did not result in any new patentable discoveries, so Gilead retained full intellectual property of its discoveries.
The investments in MCMs for VHFs were underscored by patients desiring treatment especially in austere and resource-limited locations. While the improvement of supportive care remains a priority, the need for vaccines, prevention and therapeutics remains a priority. For rare diseases with outbreak potential, lack of pre-clinical and early clinical data leads to consternation about the use of candidate drugs in patients. A delicate balance between offering hope and potential unforeseen harm caused by the candidate drug is obvious and confounding for policy makers, clinicians and patients. The 2014–2015 EVD outbreak demonstrated that there was a need to advance candidates to early phase I clinical studies to ready them for use in an actual emergency outbreak. Unfortunately, limited annual budgets focused on the traditional rare and low commercial value MCMs and emerging threats remain a challenge for the US and other countries.
Overall, the takeaways are that rare events such as pandemics of public health emergencies are prioritised and demonstrate clear markets and defined customers. Common systems for infectious disease and biothreat detection are agnostic and result in cost savings. One common trait among our case histories is that each company was ‘all-in’ when partnering with the USG. As the biotech market becomes more segmented as in information technology, companies may resort to alternative partnering models which may use technology licencing that allows multiple entities to bring products to market. The USG will need to continue providing funding incentives to maintain enterprise especially during normal peacetime. The COVID-19 pandemic demonstrates that the biotechnology sector is the core of bioeconomy in some respects and lessons learned will apply to grow, nurture and sustain the core of our overall economy, national health security and well-being.
Acknowledgments
This article was developed without any external funding. The authors thank the reviewers and editors for their critique. Special thanks to Dr. Eric Midboe for his expert insights and guidance. We also appreciate Mr Garrett Dalton’s graphic artwork.
Disclosure statement
No potential conflict of interest was reported by the author(s). This content represents the views of the author(s) only, and does not necessarily represent the views or professional advice of KPMG LLP.
Additional information
Notes on contributors
Kenneth B. Yeh
Kenneth B. Yeh is a senior director at MRIGlobal, which is an independent nonprofit research organization. He has created partnerships and reinforced best practices to develop technical solutions in the areas of biodefense, biotechnology, and global health security. Kenny’s work spans industry experience for detecting and identifying biological agents in the field to fielding cooperative biological research studies among global partners. His related publications include panel topics on end users and first movers in biotechnology, sharing of data and sample material, and research in high-containment facilities. Kenny has a background in microbiology.
Eric Du
Eric Du is a manager at KPMG’s Healthcare & Life Sciences Strategy group, which supports organic and inorganic growth strategy, merger & acquisition due diligence, integration, and performance transformation for corporate and private equity clients. Eric is experienced in R&D strategy and operations, government policy, and public health. His related publications specialize in improving U.S. defense industrial base through policy reform and public-private partnership with a focus on the biotech sector.
Gene Olinger
Gene G. Olinger is a chief science advisor at MRIGlobal in the Life Sciences Division. He is a seasoned scientist with 20+ years of experience leading complex in vitro and in vivo studies in BSL-3/BSL-4 laboratories to drive delivery of vaccines, clinical diagnostics, and countermeasure developments to emerging pathogens. As a subject matter expert in immunology, pathogenesis, host-pathogen interactions, virology, and vaccinology, he has been engaged in developing partnerships to better the health globally. He is a thought leadership is evidenced by multiple patents, 100+ peer-reviewed published manuscripts, abstracts, and reviews. Gene also serves various adjunct faculty positions with various universities.
Donna Boston
Donna S. Boston led programs for over three decades with the U.S. Government, fielding successful FDA-approved products that met the government’s medical countermeasure program requirements. Her expertise in advancing biotech/biomedical products and technologies allowed them to emerge from the development pipeline and reach the clinical market to protect our national health security. In 2018, she co-launched the BARDA DRIVe program to address gaps in preparedness and areas within the continuum of public health response that require innovative and entrepreneurial approaches. Under the pandemic task force's 2020 Operation Warp Speed, her programs resulted in the unprecedented delivery of much-needed FDA-authorized COVID-19 diagnostic tests that began shipping to labs nationwide within weeks of their development.
References
- Ardizzone, K., & Love, J. (2020). Other transaction agreements: Government contracts that eliminate protections for the public on pricing, access and competition, including in connection with COVID-19 vaccines and treatments. https://www.keionline.org/wp-content/uploads/KEI-Briefing-OTA-29june2020.pdf
- DiEuliis, D., Terrell, P., & Emanuel, P. 2020. Breaching the department of defense's biotech bottleneck. Health Security.18:2. 139–144.
- Du, E. (2020). Biotech acquisitions a different ballgame for defense department. National Defense.
- Evens, R.P. (2016). Pharma Success in Product Development—Does Biotechnology Change the Paradigm in Product Development and Attrition. The AAPS Journal, 18(1), 281–285. https://doi.org/10.1208/s12248-015-9833-6
- Government Accountability Office. (2020). Defense science and technology: Opportunities to Better Integrate Industry Independent Research and Development into DOD Planning. US Government Printing Office.
- Government Accountability Office. (2021). Information on federal contributions to remdesivir. US Government Printing Office.
- McCormick, R. (2020, December). Department of defense other transaction authority trends. Center for Strategic and International Studies.
- National Academies of Sciences, Engineering, and Medicine. (2020). National Academies of Sciences, Engineering, and Medicine. The National Academies Press. https://doi.org/10.17226/25525
- National Academy of Sciences. (2020). Engineering, and medicine. Safeguarding the bioeconomy. The National Academies Press. Retrieved July 13, 2020, from https://doi.org/10.17226/25525
- Peeler, D. (2016). Biotech business lessons for defense acquisition. Strategic Studies Quarterly.
- Sargent, Jr J. (2020). Department of defense research, development, test, and evaluation (RDT and E): appropriations structure. Congressional Research SVC Washington United States.
- Smith, E., & Diggans, J. (2020, July/August). Next Steps to Grow the Bioeconomy. Health Secur, 18 (4), 297–302. PMID: 32816592 https://doi.org/10.1089/hs.2020.0012
- Tromberg, B.J., Schwetz, T.A., Pérez-Stable, E.J., Hodes, R.J., Woychik, R.P., Bright, R.A., Fleurence, R.L., & Collins, F.S. (2020, September 10). Rapid scaling up of Covid-19 diagnostic testing in the United States—the NIH RADx initiative. New England Journal of Medicine, 383(11), 1071–1077. https://doi.org/10.1056/NEJMsr2022263
- Tsai W, and Erickson S. (2006). Early-stage biotech companies: strategies for survival and growth. Biotechnology healthcare, 3(3), 49.
- Watson, C., Watson, M., Gastfriend, D., & Sell, T.K. (2018, October 1). Federal funding for health security in FY2019. Health Security, 16(5), 281–303. https://doi.org/10.1089/hs.2018.0077
- Weaver, J.M. (2018). The 2017 National Security Strategy of the United States. Journal of Strategic Security, 11 (1), 62–71. https://doi.org/10.5038/1944-0472.11.1.1655.
- Weaver, John M. (2018). The 2017 National Security Strategy of the United States. Journal of Strategic Security, 11(1), 62–71. https://scholarcommons.usf.edu/jss/vol11/iss1/5
- Whitehouse, C.A., Bavari, S., & Perkins, M.D. (2015, October). United States FDA’s emergency use authorization of Ebola virus diagnostics: Current impact and lessons for the future. Expert Review of Molecular Diagnostics, 15(10), 1231–1235. https://doi.org/10.1586/14737159.2015.1077117
- Wilson, S.A. (2006). A novel approach for the development and acquisition of a diagnostic medical device from concept to fielding: The Joint Biological Agent Identification and Diagnostic System (JBAIDS). George Mason University.