
Abstract
Damselflies of the genus Ischnura emerge as organisms with high potential in ecological, evolutionary and developmental research at the base of flying insects. Ischnura elegans and Ischnura hastata are for example one of the few odonate species where a complete life cycle over generations can be reared under laboratory conditions. We here report the complete mitochondrial genome of Ischnura elegans as a valuable genomic resource for future eco-evo-devo studies at the base of flying insects. The genome has a total length of 15,962 bp and displays all typical features of Odonata (dragonflies and damselflies) mitochondrial genomes in gene content and order as well as A + T content. Start and stop codons of all protein-coding genes are consistent. Most interestingly, we found four intergenic spacer regions and a long A + T rich (control) region of 1196 bp, which is almost double the size of the close relative Ischnura pumilio. We assume that the adequate insert size and iterative mapping may be more efficient in assembling this duplicated and repetitive region.
The blue-tailed damselfly Ischnura elegans is a small, widely distributed European damselfly of the family Coenagrionidae. The females of this species exhibit a color polymorphism with three different color morphs (e.g. Andrés et al. Citation2000), which put them in the center of research concerning the evolution of color polymorphism (e.g. Hammers & Van Gossum Citation2008). Furthermore, the complete life cycle of this species can be cultured in the lab bridging the gaps between developmental, environmental and evolutionary studies (Simon & Hadrys Citation2013, Citation2014). It was the first odonate species for which genomic data in terms of ESTs (Simon et al. Citation2009) and a transcriptome (Chauhan et al. Citation2014) were available. A complete genome would facilitate state of the art future studies in eco-evo-devo. In a first attempt, we here present the assembly of the mitochondrial genome of I. elegans.
Total genomic DNA was extracted from the flight muscles of a single individual using a standard Phenol-Chloroform extraction (Hadrys et al. Citation1992). The specimen was collected in Schapen, northern Germany (52°16′7.95″N, 10°31′36.37″E). The library preparation and whole genome sequencing was conducted at Yale University in the Center for Genome Analyses (YCGA, http://www.ycga.yale.edu) on an Illumina HiSeq2000 (Illumina Inc.) platform generating 75 bp paired-end reads with an insert size of ∼450 bp. For the assembly of the complete mitochondrial genome Geneious vers. 8.1.5 (http://www.geneious.com) was applied as follows: one published mitochondrial gene sequence served as seed (cox2, KC430130), and a fraction of the cleaned reads were mapped onto the seed using iterative mapping. Hereby the iterations were increased with length of the seed sequence (from 5 to 25) as well as the overlap identity, which was initially placed at 90%. A maximum overlap of 45–50 bp was chosen to prevent miss-mapping. In addition, the length of the A + T rich (control) region was confirmed via PCR. The continuous annotation was conducted using the MITOS WebServer (mitos.bioinf.uni-leipzig.de/index.py) and verified via BLAST (Altschul et al. Citation1990) against GenBank, already published mitochondrial genomes (e.g. Tang et al. Citation2014; Yu et al. Citation2014; Chen et al. Citation2015) and especially against the closest relative Ischnura pumilio (NC_021617; Lorenzo-Carballa et al. Citation2014). Transfer RNAs were predicted using the tRNAscan-SE vers.1.21 Search Server (http://lowelab.ucsc.edu/tRNAscan-SE; Lowe & Eddy Citation1997) and ARWEN vers. 1.2 (http://mbio-serv2.mbioekol.lu.se/ARWEN; Laslett & Canbäck Citation2008). Finally, a phylogeny was reconstructed using four other selected Odonata species and I. elegans (). Based on a concatenated alignment of all 13 protein-coding genes and the rRNA genes a maximum parsimony tree was calculated using PAUP vers. 4.0b10 (Swofford Citation2002) with a heuristic search under the 50% majority-rule and 1,000 bootstrap replicates.
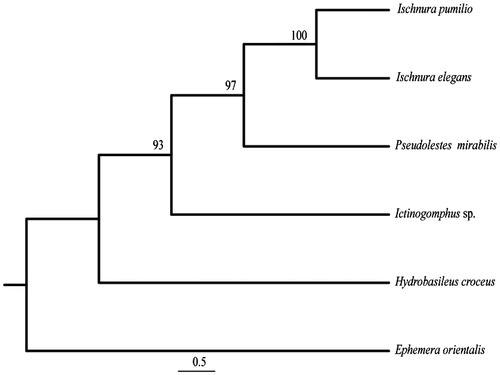
Figure 1. Phylogenetic position of I. elegans and I. pumilio (NC_021617), Pseudolestes mirabilis (NC_020636), Hydrobasileus croceus (NC_025758), Ictinogomphus sp. (KM244673) using the mayfly Ephemera orientalis (NC_012645) as outgroup. MP bootstrap supports are shown for each node. I. elegans and I. pumilio exhibit 86.4% identity in the mt-genes used.
The present mitogenome has a total length of 15,962 bp (GenBank accession number: KU958378) and is 712 bp longer than I. pumilio’s with a high overall similarity of 86.4%. It displays the typical metazoan gene content with 13 protein coding genes (PCGs), 2 rRNA genes (16S and 12S) and 22 tRNA genes with an identical gene order to all other odonate mitogenomes published to date (). Furthermore, four intergenic spacer regions were detected which are consistent in position with I. pumilio and Megaloprepus caerulatus (Lorenzo-Carballa et al. Citation2014; Feindt et al. Citation2016) but differ in size. Base composition of the I. elegans mitogenome is AT biased (A: 40.3%, T: 32.5%, G: 12.3%, C: 14.8%) so as all protein coding genes (71.4%), rRNAs (75.1%) and tRNAs (72.1%) on average. Except for cox1 (TTA) all PCGs initiate with standard invertebrate mitochondrial start codons: cox3, nad4, nad4L and cytb with ATG; nad2, cox2, nad5, with ATT; atp8, atp6, nad3, nad1 with TTG; and nad6 with ATC. Complete stop codons terminate nine genes (TAA: nad2, cox1, atp8, atp6, nad4L, nad6, cytb, nad1; TAG: nad3) whereas four proteins use incomplete stop codons with post-transcriptional polyadenylation (cox2, cox3, nad5, nad4). Transfer RNAs vary in size from 65 to 72 bp and all of them fold into the characteristic clover-leaf secondary structure. Overlapping gene junctions were observed for 13 genes, the longest overlap between atp6 and atp8 is 13 bp.
Table 1. Organization of the mitochondrial genome of Ischnura elegans.
The different length of the presented mt genome compared to I. pumilio is mainly based on the almost two times longer A + T rich (control) region. Since we proved the length of the A + T rich (control) region via PCR, we assume that the combination of an appropriate insert size and iterative mapping may be more accurate for the assembly of long repetitive and duplicated regions. These usually tend to challenge genome assembly software. The control region comprises a triplicated motive of in total almost 600 bp, which could only be resolved correctly with consideration to the insert size. This could be an important aspect for future library preparation on Illumina platforms.
Disclosure statement
The authors declare no conflict of interest to other working groups.
Funding
This work was supported by a travel grant of the Graduate Academy of Leibniz University Hannover given to WF and a fellowship of the German Academic Exchange Service given to RH (PROMOS, DAAD). HJO acknowledges a doctoral fellowship of the Studienstiftung des deutschen Volkes.
References
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol. 215:403–410.
- Andrés JA, Sánchez-Guillén RA, Cordero Rivera A. 2000. Molecular evidence for selection on female color polymorphism in the damselfly Ischnura graellsii. Evolution. 54:2156–2161.
- Chauhan P, Hansson B, Kraaijeveld K, de Knijff P, Svensson EI, Wellenreuther M. 2014. De novo transcriptome of Ischnura elegans provides insights into sensory biology, colour and vision genes. BMC Genomics. 15:1.
- Chen M-Y, Chaw S-M, Wang J-F, Villanueva RJT, Nuneza OM, Lin C-P. 2015. Mitochondrial genome of a flashwing demoiselle, Vestalis melania from the Philippine Archipelago. Mitochondrial DNA. 26:720–721.
- Feindt W, Osigus H-J, Herzog R, Mason CE, Hadrys H. 2016. The complete mitochondrial genome of the neotropical helicopter damselfly Megaloprepus caerulatus (Odonata: Zygoptera) assembled from next generation sequencing data. Mitochondrial DNA. [Epub ahead of print]. Doi: http://dx.doi.org/10.1080/23802359.2016.1192504.
- Hadrys H, Balick M, Schierwater B. 1992. Applications of random amplified polymorphic DNA (RAPD) in molecular ecology. Mol Ecol. 1:55–63.
- Hammers M, Van Gossum H. 2008. Variation in female morph frequencies and mating frequencies: random, frequency-dependent harassment or male mimicry? Animal Behav. 76:1403–1410.
- Laslett D, Canbäck B. 2008. ARWEN: a program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics. 24:172–175.
- Lorenzo-Carballa MO, Thompson DJ, Cordero-Rivera A, Watts PC. 2014. Next generation sequencing yields the complete mitochondrial genome of the scarce blue-tailed damselfly, Ischnura pumilio. Mitochondrial DNA. 25:247–248.
- Lowe TM, Eddy SR. 1997. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25:955–964.
- Rivera AC, Sánchez-Guillén RA. 2007. Male-like females of a damselfly are not preferred by males even if they are the majority morph. Animal Behav. 74:247–252.
- Simon S, Strauss S, von Haeseler A, Hadrys H. 2009. A phylogenomic approach to resolve the basal pterygote divergence. Mol Biol Evol. 26:2719–2730.
- Simon S, Hadrys H. 2013. A comparative analysis of complete mitochondrial genomes among Hexapoda. Mol Phylogen Evol. 69:393–403.
- Simon S, Hadrys H. 2014. Phylogeny of the most species rich group on Earth, the Pterygota: ancient problems, living hypotheses and bridging gaps, In: Wägele JW, Bartholomaeus T, editors. Deep metazoan phylogeny: the backbone of the tree of life: new insights from analyses of molecules, morphology, and theory of data analysis. Berlin: Walter de Gruyter GmbH. p. 343–359.
- Swofford DL. 2002 PAUP* phylogenetic analysis using parsimony (*and other methods). Sunderland: Sinauer Associates.
- Tang M, Tan M, Meng G, Yang S, Su X, Liu S, Song W, Li Y, Wu Q, Zhang A. 2014 . Multiplex sequencing of pooled mitochondrial genomes-a crucial step toward biodiversity analysis using mito-metagenomics. Nucleic Acids Res. 42:e166.
- Yu P, Cheng X, Ma Y, Yu D, Zhang J. 2014. The complete mitochondrial genome of Brachythemis contaminata (Odonata: Libellulidae). Mitochondrial DNA. 27:1–2.