
Abstract
In this study, the complete mitochondrial genome of Acanthurus leucosternon was determined. The genome is circular, double-stranded DNA molecule with 16,434 bp in length (EU 136032). The structure, gene organization, and gene order is similar as in other vertebrates. The overall base composition is A (29.29%), C (28.53%), T (26.37%), G (15.81%), and with 44.33% G + C content. Overlapping observed between contiguous genes is encoded in the opposite strands. The two 12S and 16S rRNA genes consist of 949 and 1690 nucleotides, respectively. Non-coding control region is 754 bp long and origin of replication is located in the WANCY cluster. Conserved domains in the control regions and secondary structures for the OL regions are also predicted. Molecular phylogenetic analysis using Maximum-Likelihood method of A. leucosternon based on the protein-coding gene sequence was also studied.
Acanthurus leucosternon is a marine ornamental surgeon fish in the Family Acanthuridae inhabiting coral reefs and sea grass beds. They occur in most tropical and subtropical seas of the world. In India Acanthurus is found throughout the Indian Ocean.
In this present study, the complete mitochondrial genome of A. leucosternon was amplified, cloned, and sequenced. Live fishes of A. leucosternon were collected from the Bay of Bengal at Rameswaram, Tamilnadu, India. Specimens of A. leucosternon (Voucher No: AE05) were preserved in the laboratory museum. The complete circular-double-stranded mitochondrial genome contained a total of 16,434 bp (GenBank accession no EU 136032) with 2rRNAs genes, 13 protein-coding genes, 22 tRNA genes with a control region required for mitochondrial protein synthesis. Most of these genes are encoded by the H-strand, while ND6 and other eight tRNAs in the light (L) strand as in other bony fishes (Inoue et al. Citation2000). The intergenic spacers of variable lengths of 1–37 nucleotides are also found similar to salmonids (Wilhelm et al. Citation2003). Acanthurus leucosternon mitogenome constitute 70% protein-coding genes, 16rRNA, 9% tRNA, and 5% non-coding regions. The A + T content is high (55.67%) with low G (15.81%) similar to other vertebrates.
The two 12S and 16S rRNA genes consist of 949 and 1690 nucleotides, respectively. The 22 tRNA genes are dispersed along the genome range in size from 65–76 nucleotides, are predicted to fold into the expected clover leaf secondary structure. Overlapping is found between tRNAGln and tRNAMet in a single nucleotide.
Acanthurus leucosternon mitogenome have all protein-coding genes and their arrangements similar to other vertebrate mtDNA. Restricted overlapping is observed in the opposite strands between contiguous genes as in most other fishes (Broughton et al. Citation2001; Vittas et al. Citation2011), in particular ATP8-ATP6, ND4L-ND4, and ND5-ND6 share 10, 7, and 4 nucleotides, respectively.
All protein-coding genes start with ATG. ND1, ATPase 8, ND4L, and ND5 ends with TAA, ND6 with TAG and the remaining genes with incomplete TA or T residues. This is quite common and typical among mtDNA genes to be completed by post transcriptional polyadenyalation (Ojala et al. Citation1981). The overall base composition for the protein genes is 69.57%, A + T composition is 55.36%.
The OL was identified as 37bp sequence located between tRNAAsn and tRNACys in the cluster of WANCY region and sharing 14 bp at 3′ and 5′ end, respectively. The control region of A. leucosternon is 754 bp long with rich A + T (66.71%). Several conserved domains such as one termination associated sequence (TAS), one central conserved sequence block and three putative conserved sequence blocks were identified in the control region.
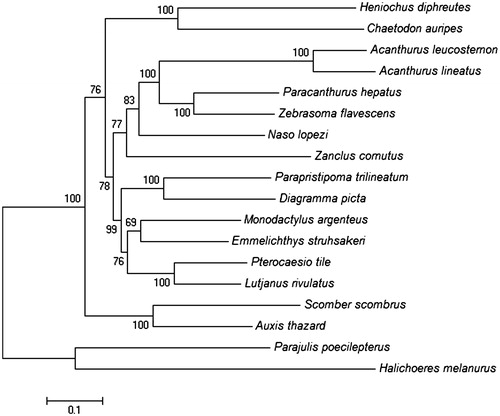
The ML tree was constructed based on the mitochondrial encoded H-strand protein genes of 18 Perciformes fishes. To find the best-fit substitution model using Modeltest version 3.6 program was employed (Posada & Crandall Citation1998). Based on the resulting ML tree (), A. leucosternon and A. lineatus are grouped together forming sister taxon to the genus Paracanthus and Zebrasoma within the order Acanthuroidei.
Figure 1. ML tree was generated using the MEGA 6 program based on GTR + I + G model. Initial tree for the heuristic search was obtained by applying the Neighbor-Joining method to a matrix of pairwise distances estimated using the Maximum-Composite Likelihood (MCL) approach. The accession numbers for all 18 species are Halichoeres melanurus (AP006018.1), Chaetodon auripes (AP006004.1), A. leucosternon (EU136032.1), A. lineatus (EU273284.2), Paracanthurus hepatus (KT826539.1), Zebrasoma flavescens (AP006032.1), Naso lopezi (AP009163.1), Zanclus cornutus (AP009162.1), Parapristipoma trilineatum (AP009168.1), Diagramma picta (AP009167.1), Monodactylus argenteus (AP009169.1), Emmelichthys struhsakeri (AP004446.1), Pterocaesio tile (AP004447.1), Lutjanus rivulatus (AP006000.1), Scomber scombrus (AB120717.1), Auxis thazard (AB105447.1), Parajulis poecilepterus (EF192032.2), and Heniochus diphreutes (AP006005.1). There were a total of 10,866 positions in the final dataset. The tree with the highest log likelihood (−102591.12) is shown. The number of nucleotide substitutions per site is indicated by the scale.
Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
Related Research Data
References
- Broughton RE, Milam JE, Roe BA. 2001. The complete sequence of the zebrafish (Danio rerio) mitochondrial genome and evolutionary patterns in vertebrate mitochondrial DNA. Genome Res. 11:1958–1967.
- Inoue JG, Miya M, Tsukamota K, Nishida M. 2000. Complete mitochondrial DNA sequence of the Japanese sardine Sardinops melanostictus. Fisheries Sci. 66:924–932.
- Ojala D, Montoya J, Attardi G. 1981. tRNA punctuation model of RNA processing in human mitochondria. Nature. 290:470–474.
- Posada D, Crandall KA. 1998. MODELTEST: testing the model of DNA substitution. Bioinformatics. 14:817–818.
- Vittas S, Drosopoulou E, Kappas I, Pantzartzi CN, Scouras ZG. 2011. The mitochondrial genome of the European catfish, Silurus glanis (Siluriformes, Siluridae). J Biol Res-Thessalon. 15:25–35.
- Wilhelm V, Villegas J, Miquel A, Engel E, Bernales S, Valenzuela PDT, Burzio LO. 2003. The complete sequence of the mitochondrial genome of the Chinook salmon, Oncorhynchus tshawytscha. Biol Res. 36:223–231.