
Abstract
The complete mitochondrial genome sequence was determined for a specimen of Calotomus japonicus, a temperate parrotfish endemic to coastal East Asia. It was compared phylogenetically with previously published partial sequences from this species and other parrotfishes. The obtained tree indicated that the three cytb sequences of C. japonicus from a recent molecular study (LC068806-8) probably resulted from introgression through intergeneric hybridization, or possibly from sample confusion. Taking the presently obtained mitogenome as representative of C. japonicus, the species most closely related to this one among congeners is C. zonarchus, which is endemic to the Hawaiian islands.
Keywords:
Calotomus japonicus, which belongs to the family Scaridae, is a temperate parrotfish endemic to East Asian coastal waters. Within this mainly tropical family, only this species and Scarus ovifrons are common in the waters of southern Japan (Kishimoto Citation1988). Although the consumption of S. ovifrons sometimes causes poisoning in humans (Suzuki et al. Citation2013), C. japonicus is an important target species for local fisheries (Kume et al. Citation2010). These two species belong to different genera and are easily distinguished by their morphological features (Shimada Citation2013). A recent molecular study by Ogawa et al. (Citation2015), however, reported that 477-base-pair (bp) fragments of the mitochondrially encoded cytochrome b (cytb) gene were almost identical between them, and further that the cytb haplotypes from C. japonicus were clustered not into the clade of Calotomus, but into that of Scarus. Considering their taxonomic status, I suspected that the cytb sequences of C. japonicus reported by the authors might not be representative of this species. To investigate this possibility, I determined the complete mitochondrial DNA sequence of C. japonicus, and compared it phylogenetically with the cytb sequences reported by Ogawa et al. (Citation2015) and other previously published sequences of parrotfishes.
The complete mitochondrial genome (mitogenome) of C. japonicus was determined by a polymerase chain reaction (PCR)-based technique (Mabuchi et al. Citation2004) using a specimen purchased at Odawara Fish Market in Kanagawa Prefecture, Japan, and deposited at the National Science Museum, Tokyo (registered number NSMT-P 65394). The obtained mitogenome (DDBJ accession no. AP017568) is 17,199 bp in length, which is ca. 500–520 bp longer than the five parrotfish mitogenomes reported thus far (). Most of the differences in length result from variation in the length of intergenic regions: the control region of C. japonicus is longer than those of the five parrotfishes by ca. 400 bp, and the intergenic region between tRNASer and tRNALeu of the species is ca. 30 bp longer than those of the others (). As already reported by Mabuchi et al. (Citation2004), this species shares a gene rearrangement accompanied by a tRNA pseudogene with other parrotfishes. In the typical gene order of vertebrates, the tRNA gene cluster between the ND1 and ND2 genes includes the tRNAIle (I), tRNAGln (Q), and tRNAMet (M) genes in this order (IQM). However, in the parrotfish mitogenomes, the tRNAMet gene is inserted between the tRNAIle and tRNAGln genes, and the tRNAGln gene is followed by a putative tRNAMet pseudogene (ΨM) (making the gene/pseudogene order IMQΨM).
Table 1. Sequence lengths (base pairs) of whole and major intergenic regions of six parrotfish mitogenomes.
Using the six mitogenomes and other available partial sequences of parrotfishes, a phylogenetic tree was constructed using a supermatrix approach (de Queiroz & Gatesy Citation2007) by the method described in the legend of . The resultant supermatrix tree recovered the two major parrotfish clades of Streelman et al. (Citation2002): one was the “Seagrass” clade including Cryptotomus, Nicholsina, and Sparisoma, as well as Calotomus, and the other was the “Reef” clade including Scarus and Chlorurus, although the phylogenetic position of Leptoscarus differed from that in the previous study, in which this genus was included in the “Seagrass” clade. The presently obtained mitogenome of C. japonicus was nested within the “Seagrass” clade, and formed a Calotomus clade together with a partial 12S rRNA sequence of this species and the cytb and COI sequences of the other congeneric species. The cytb sequences of C. japonicus reported by Ogawa et al. (Citation2015) (indicated by arrows) were, on the other hand, included in the “Reef” clade, and showed a very close relationship with the cytb sequences of S. ovifrons, as shown in Supplementary Material 1 in the report by Ogawa et al. (Citation2015). Based on these results, I concluded that the presently obtained mitogenome of C. japonicus is more representative of this species than the cytb sequences of C. japonicus reported by Ogawa et al. (Citation2015). The latter sequences (LC068806-8) probably resulted from introgression through intergeneric hybridization between C. japonicus and S. ovifrons, or possibly from sample confusion.
Figure 1. Supermatrix tree of 24 selected mitochondrial DNA sequences of parrotfishes with a labrid fish, Cheilinus undulatus, as an outgroup. Accession numbers are indicated after the species names with gene regions and their lengths in parentheses for partial sequences. Bootstrap support (70% or over) is indicated at the nodes. For the three arrows, see the text. The tree backbone indicated by bold lines was first generated for the boxed species (for which mitogenome sequences were available) by partitioned maximum-likelihood (ML) analysis using 13 protein-coding and two ribosomal RNA gene sequences, as follows. Multiple alignment was conducted for each of the 15 genes using the online version of MAFFT (http://mafft.cbrc.jp/alignment/server/), and ambiguous regions were trimmed using trimAl (ver. 1.2; Capella-Gutiérrez et al. Citation2009) with the “strict” setting. For the concatenated 13,751-bp dataset, the optimal partition model was determined using PartitionFinder (ver. 1.1.1; Lanfear et al. Citation2012) with codons of each protein-coding gene considered separately. Using the selected model including 10 partition subsets, ML analysis was performed using RAxML (ver. 8.1.5; Stamatakis Citation2014) with the following settings: -f a; -m GTRGAMMA; -# 1000. The obtained ML tree was then used as a backbone constraint for the supermatrix tree. The supermatrix tree was constructed based on the dataset including the 25 sequences, which were prepared by the same methods as for the backbone tree. For the resultant 13,758-bp dataset, ML analysis was performed by RAxML using the -r option and the same partition model as for the backbone tree, with the other settings as follows: -f a; -m GTRCAT; -# 1000; -V.
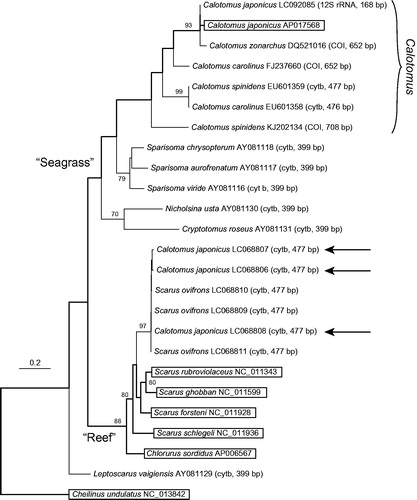
The presently obtained mitogenome of C. japonicus revealed an interesting biogeographic pattern within the genus Calotomus. This genus consists of five species (Randall Citation1985), and the present dataset included four of the five (only Calotomus viridescens, endemic to the Red Sea, was absent). Based on the present supermatrix tree (), the species most closely related to C. japonicus, which is endemic to coastal East Asia, is neither C. carolinus nor C. spinidens, both of which are widely distributed in the Indo-West Pacific region, but a species geographically isolated from C. japonicus, C. zonarchus, endemic to the Hawaiian islands.
Acknowledgments
I thank Drs. Keiichi Matsuura, Gento Shinohara, Masanori Nakae and Kaoru Kuriiwa (National Museum of Nature and Science, Tokyo) for registering the specimens in the NSMT fish collection and helping me to examine them.
Disclosure statement
The author alone is responsible for the content and writing of this paper.
Funding
This work was supported by CREST “Development of marine ecosystem evaluation methods in the high throughput sequencing era” funded by Japan Science and Technology Agency.
References
- Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T. 2009. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 25:1972–1973.
- de Queiroz A, Gatesy J. 2007. The supermatrix approach to systematics. Trends Ecol Evol (Amst). 22:34–41.
- Kishimoto H. 1988. Family Scaridae. In: Masuda H, Amaoka K, Araga C, Uyeno T, Yoshino T, editors. The fishes of the Japanese Archipelago. 2nd ed. Tokyo: Tokai University Press; p. 213–221.
- Kume G, Kubo Y, Yoshimura T, Kiriyama T, Yomaguchi A. 2010. Life history characteristics of the protogynous parrotfish Calotomus japonicus from northwest Kyusyu, Japan. Ichthyol Res. 57:113–120.
- Lanfear R, Calcott B, Ho SY, Guindon S. 2012. Partitionfinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol Biol Evol. 29:1695–1701.
- Mabuchi K, Miya M, Satoh TP, Westneat MW, Nishida M. 2004. Gene rearrangement and evolution of tRNA pseudogenes in the mitochondrial genome of the parrotfish (Teleostei: Perciformes: Scaridae). J Mol Evol. 59:287–297.
- Ogawa N, Seki S, Yamada H, Nakamura Y. 2015. Development of a Japanese parrotfish species identification method using PCR-RFLP analysis and its application – determination of the species composition of parrotfish juveniles in seagrass beds of the Ryukyu Islands, southern Japan. Aquacult Sci. 63:423–435.
- Randall JE. 1985. A revision of the Indo-West Pacific parrotfish genera Calotomus and Leptoscarus (Scaridae: Sparisomatinae). Indo-Pacific Fishes. 5:1–32.
- Shimada K. 2013. Scaridae. In: Nakabo T, editor. Fishes of Japan with pictorial keys to the species. 3rd ed. Kanagawa: Tokai University Press; p. 1137–1151.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30:1312–1313.
- Streelman JT, Alfalo M, Westneat MW, Bellwood DR, Karl SA. 2002. Evolutionary history of the parrotfishes: biogeography, ecomorphology, and comparative diversity. Evolution. 56:961–971.
- Suzuki T, Watanabe R, Matsushima R, Ishihara K, Uchida H, Kikutsugi S, Harada T, Nagai H, Adachi M, Yasumoto T, Murata M. 2013. LC-MS/MS analysis of palytoxin analogues in blue humphead parrotfish Scarus ovifrons causing human poisoning in Japan. Food Addit Contam Part A Chem Anal Control Expo Risk Assess. 30:1358–1364.