
Abstract
We have sequenced the female and male mtDNA of Unio delphinus and inferred the Unionidae phylogeny using 41 complete mtDNA sequences. Additionally, we compared the concatenated mtDNA trees with those using single or combination of two mtDNA genes to identify the best genes to use in the absence of complete mitogenomes. The gender-specific mtDNAs of U. delphinus contain all Unionida mtDNA specific features. The mtDNA phylogeny supports the reciprocal monophyly of the gender-specific clades but it was inconclusive regarding Unionidae subfamilies relationships. The gene trees topologies using ND5 or 16S-rRNA with ND1 were the closest trees to the mtDNA trees.
The genus Unio is among the most widespread freshwater mussel taxa in the world (Bogan & Roe Citation2008). However, the recently redescribed Unio delphinus (Froufe et al. Citation2016a; Lopes-Lima et al. Citation2016) is restricted to the main Atlantic basins of the Iberian Peninsula (Froufe et al. Citation2016a). This species was considered as ‘Near Threatened’ in the last IUCN Red List assessment, making its genetic characterization a priority for conservation (Cuttelod et al. Citation2011; Lopes-Lima et al. Citation2014). All Unionidae species have an interesting mitochondrial inheritance mechanism known as Doubly Uniparental Inheritance (DUI). In this process, all individuals possess the maternally transmitted mtDNA (F-type), but the males have also a paternally inherited mtDNA, which is found primarily in sperm mitochondria of male gonads (M-type) (Zouros et al. Citation1994). Phylogenetic studies using the M-type mtDNA are rare, due to the lack of available sequences. Thus, it is not surprising that M-type mitogenomes have never been sequenced for the Unioninae subfamily. Of the few published phylogenies using the whole mtDNA within the Unionida, only two of the six families are represented, Unionidae and Margaritiferidae (e.g. Huang et al. Citation2013; Froufe et al. Citation2016b). Therefore, most of the published Unionida phylogenies use a combination of two out of three mtDNA genes: Cytochrome c oxidase subunit 1 (COX1), 16S ribosomal RNA (16S-rRNA) and NADH ubiquinone oxidoreductase core subunit 1 (ND1) (e.g. Graf & Cummings Citation2006; Whelan et al. Citation2011; Campbell & Lydeard Citation2012; Prié & Puillandre Citation2014). Curiously, no studies have been published so far, exploring which of these gene combinations best represent the concatenated mtDNA phylogeny. On the other hand, the utility of the remaining mitochondrial regions has yet to be revealed. The main objectives of this study are to (i) sequence and characterize the gender-specific mtDNA for the U. delphinus; (ii) infer the phylogenetic relationships among Unionida species using both the F- and M-type mtDNA sequences; and (iii) determine the mtDNA gene and combination of two genes that best represent the mtDNA phylogeny.
Living specimens (n = 3) of U. delphinus were collected from the Barrinha de Mira Lagoon, Vouga River Basin (Portugal, geospatial coordinates: 40.450047; −8.797070). Dissected gonadal and mantle tissues for one male individual (sample code UDE006) were selected for DNA extractions (Jetquick tissue DNA Spin Kit – Genomed). The Ion Xpress Plus Fragment Library Preparation Kit (Life Technologies, Carlsbad, CA) was used to prepare the genomic DNA shotgun library. All sequencing was performed on the Ion Torrent PGM using the Ion PGMTM 200 and 400 Sequencing kit and 316 and 318 semiconductor chips following manufacturer recommendations. The mtDNA genomes were assembled de novo into contigs using CLC bio’s Genomics Workbench (GW; version 7.0) software. Gene annotations were performed using MITOS (Bernt et al. Citation2013) and the gene limits were then checked using BLASTX (Altschul et al. Citation1997). Putative origins of replication were identified using the approach described in Fonseca et al. (Citation2014). For the phylogenitic analyses, the two U. delphinus haplotypes sequenced were used together with 39 Unionida available mitogenomes (list of genomes and respective accession numbers used supplied on request). The DNA and amino acids (AA) sequences of all mtDNA protein-coding genes (PCG), except ATP8 and the gender-specific open reading frames (M-ORF, H-ORF, and F-ORF) were used. The sequences of each gene were aligned using GUIDANCE (version 1.5, Penn et al. Citation2010; see Froufe et al. Citation2016b for the parameters used). The gene alignments were concatenated, resulting in two alignments with the following composition: 11,865 nucleotides or 3962 amino acids. The best-fit models of DNA and protein evolution were selected using jModeltest 2 (Darriba et al. Citation2012) and ProtTest 3.3 (Darriba et al. Citation2011), respectively. The Maximum-Likelihood (ML) phylogenetic inference was performed using RAxML (version 8.0.0, Stamatakis Citation2014) with 100 rapid bootstrap replicates and 20 ML searches. The Bayesian methodology was applied using MrBayes v3.2.1 (Ronquist et al. Citation2012) with two independent runs (1 × 107 generations with a sampling frequency of 1 tree for every 100 generations), each with four chains (3 hot and 1 cold). All runs reached convergence (average standard deviation of split frequencies below 0.01). The posterior distribution of trees was summarized in a 50% majority rule consensus tree (burn-in of 25%). The topological differences between trees were measured using a normalization of the Robinson and Foulds distance metric (nRF-distance, Robinson & Foulds Citation1981) implemented in RAxML.
The length of the female (15,762 bp; NCBI accession number KT326917) and male (16,620 bp; NCBI accession number KT326918) mitogenomes of U. delphinus is within the expected range for each gender-specific haplotype within Unionida (data supplied on request). The M-type genome is larger because the protein-coding genes COX2 and M-ORF are also larger. Both haplotypes have the 13 protein-coding genes typically found in metazoan mitochondrial genomes, the gender-specific ORF described for all Unionida mitogenomes with DUI system (Breton et al. Citation2009, Citation2011), 22 transfer RNA (tRNA), and 2 ribosomal RNA (rRNA) genes. The gene orientation, but not gene order, is the same in both mtDNA haplotypes (genomic details supplied on request). The most likely control-region, among all predicted in this study (data supplied on request) is located after ND5 (plus tRNA-H in the M-type) and before the cluster of tRNA genes (tRNA-Q, tRNA-C, tRNA-I, tRNA-V, and tRNA-L) as: (i) it is among the largest non-coding regions, (ii) it is shared by both gender-specific U. delphinus mitogenomes, (iii) it contains sequences with the ability to form stable hairpin structures and direct repeats (F-type), (iv) which are located in a region where the AT proportion is among the highest (AT% around 68%). Additionally, this region was identified as the control-region in other unionid mtDNAs (Breton et al. Citation2009; Huang et al. Citation2013).
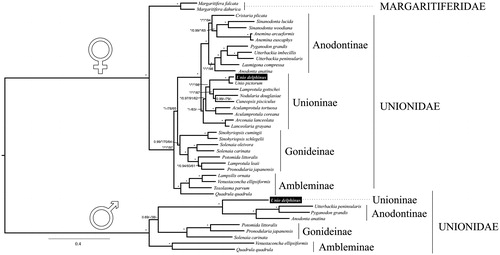
All the phylogenies inferred in this study support the reciprocal monophyly of the Unionidae + Margaritiferidae female lineages and the monophyly of the Unionidae M-type (; all phylogenetic trees figures supplied on request). They also support the F-clade monophyly of the subfamilies Anodontinae and Ambleminae as well as all M-clade subfamilies present in this study (). The subfamily Unioninae was recovered with confidence only in the BI-DNA tree. The F-type mtDNA of U. delphinus was sister taxon of U. pictorum in all analyses with maximum support. The Gonideinae subfamily was monophyletic in all trees, with high support in the BI trees only. Regarding subfamilies relationships, the DNA trees and the F-clade of the ML-AA tree support the topology (Ambleminae (Gonideinae (Unioninae + Anodontinae))), whereas the BI-AA tree and the M-clade of the ML-AA tree support the topology ((Ambleminae + Gonideinae) (Unioninae + Anodontinae)) (). In agreement with Huang et al. (Citation2013), our results indicate that using different phylogenetic methodologies (ML or BI) or sequence data types (DNA or amino acids) may result in distinct mitogenomic trees. This might be due to deficient and/or biased sampling, which could be corrected with the inclusion of more and widely distributed taxa representing all Unionidae subfamilies. In addition, future phylogenetic studies should also use nuclear markers as well as concatenated full M- with F-mitogenomes.
Figure 1. Phylogenetic BI tree of Unionida (freshwater mussels) estimated from 12 concatenated individual mitochondrial nucleotide gene sequences. The phylogenetic tree was inferred using MrBayes (version 3.2.1). The values for branch support are represented in the following order: (1) The Bayesian posterior probabilities for BI-DNA tree, (2) The Bayesian posterior probabilities for BI-AA tree, (3) ML bootstrap support values for ML-DNA, (4) ML bootstrap support values for ML-AA tree, (3), (4). Maximum supporting values (BI =1 and ML =100) are represented with ‘*’. The mitogenomes sequenced for this study are highlighted in the tree inside a box.
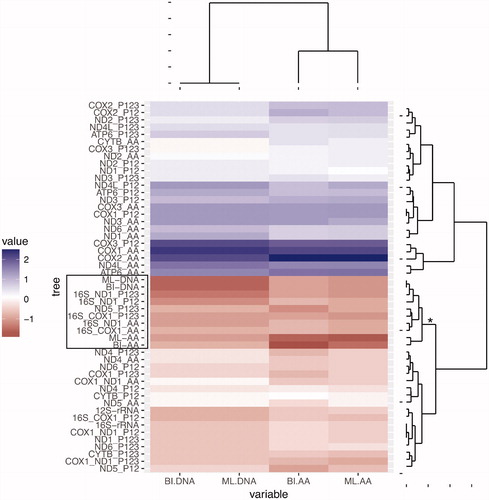
The trees using ND5 sequences were the most similar to the mtDNA trees (table with topological distances supplied on request). As for the most used single genes COX1, ND1, and 16S-rRNA, the topological differences between these trees and the mitogenome trees are similar (). Finally, when using two gene sequences concatenated, the tree topologies closer to the mtDNA trees were obtained with 16S-rRNA + ND1, followed by 16S-rRNA + COX1 ().
Figure 2. Hierarchical clustering analysis of the nRF distances between all trees inferred in this study (concatenated, single gene and two genes combined). The analysis was performed using the Euclidean distance measure and is represented by a heatmap and dendrograms. The columns of the numeric matrix were centered (by subtracting the column means of the matrix from their corresponding columns) and scaled (by dividing the (centered) columns of the matrix by their standard deviations). The matrix is divided in two major groups/clades. The clade with negative scaled values (lower major clade in the figure) groups trees with smaller nRF distance relative to the mtDNA trees (darker tones correspond to lower negative values that correspond to better trees). The clade with positive scaled values (upper major clade in the figure) groups trees with larger nRF relative to the mtDNA trees (darker tones correspond to higher values that correspond to worst trees). For all individual genes and combination of two genes, we estimated the phylogenetic tree using the amino acid sequences ?_AA?, the first two codon positions ?_P12? or all codon positions ‘_P123’. The phylogenetic analyses of the single gene or combination of two genes were performed using RAxML. The clade that includes all four concatenated trees together with the best single gene tree and combination of two genes are highlighted: (i) gene names are delimited by a box in the heatmap; (ii) in the right side dendrogram, the branch leading to the clade contains an asterisk.
Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article. Any use of trade, product, or firm names is for descriptive purposes only and does not imply endorsement by the U.S. Government.
Related Research Data
References
- Altschul S, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389–3402.
- Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G, Pütz J, Middendorf M, Stadler PF. 2013. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 69:313–319.
- Bogan AE, Roe KJ. 2008. Freshwater bivalve (Unioniformes) diversity, systematics, and evolution: status and future directions. J N Am Benthol Soc. 27:349–369.
- Breton S, Beaupre HD, Stewart DT, Piontkivska H, Karmakar M, Bogan AE, Blier PU, Hoeh WR. 2009. Comparative mitochondrial genomics of freshwater mussels (Bivalvia: Unionoida) with doubly uniparental inheritance of mtDNA: gender-specific open reading frames and putative origins of replication. Genetics. 183:1575–1589.
- Breton S, Ghiselli F, Passamonti M, Milani L, Stewart DT, Hoeh WR. 2011. Evidence for a fourteenth mtDNA-encoded protein in the female-transmitted mtDNA of marine mussels (Bivalvia: Mytilidae). PLoS One. 6:e19365. doi: 10.1371/journal.pone.0019365.
- Campbell DC, Lydeard C. 2012. Molecular systematics of Fusconaia (Bivalvia: Unionidae: Ambleminae). Am Malacol Bull. 30:1–17.
- Cuttelod A, Seddon M, Neubert E. 2011. European Red List of non-marine molluscs. Luxembourg: Publications Office of the European Union.
- Darriba D, Taboada GL, Doallo R, Posada D. 2011. ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics. 27:1164–1165.
- Darriba D, Taboada GL, Doallo R, Posada D. 2012. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods. 9:772. doi: 10.1038/nmeth.2109.
- Fonseca MM, Harris DJ, Posada D. 2014. The inversion of the control region in three mitogenomes provides further evidence for an asymmetric model of vertebrate mtDNA replication. PLoS One. 9:e106654. doi: 10.1371/journal.pone.0106654.
- Froufe E, Gonçalves DV, Teixeira A, Sousa R, Varandas S, Ghamizi M, Zieritz A, Lopes-Lima M 2016a. Who lives where? Molecular and morphometric analyses clarify which Unio species (Unionida, Mollusca) inhabit the southwestern Palearctic. Org Divers Evol. 16:597. doi: 10.1007/s13127-016-0262-x.
- Froufe E, Gan HM, Lee YP, Carneiro J, Varandas S, Teixeira A, Zieritz A, Sousa R, Lopes-Lima M. 2016b. The male and female complete mitochondrial genome sequences of the endangered freshwater mussel Potomida littoralis (Cuvier, 1798) (Bivalvia: Unionidae). Mitochondrial DNA Part A. 27:3571–3572.
- Graf DL, Cummings KS 2006. Palaeoheterodont diversity (Mollusca: Trigonioida + Unionoida): what we know and what we wish we knew about freshwater mussel evolution. Zool J Linn Soc. 148:343–394.
- Huang XC, Rong J, Liu Y, Zhang MH, Wan Y, Ouyang S, Zhou CH, Wu XP. 2013. The complete maternally and paternally inherited mitochondrial genomes of the endangered freshwater mussel Solenaia carinatus (Bivalvia: Unionidae) and implications for Unionidae Taxonomy. PLoS One. 8:e84352. doi: 10.1371/journal.pone.0084352.
- Lopes-Lima M, Teixeira A, Froufe E, Lopes A, Varandas S, Sousa R. 2014. Biology and conservation of freshwater bivalves: past, present and future perspectives. Hydrobiologia. 735:1–13.
- Lopes-Lima M, Sousa R, Geist J, Aldridge DC, Araujo R, Bergengren J, Bespalaya Y, Bódis E, Burlakova L, Van Damme D, et al. Forthcoming 2016. Conservation status of freshwater mussels in Europe: state of the art and future challenges. Biol Rev. doi: 10.1111/brv.12244.
- Penn O, Privman E, Ashkenazy H, Landan G, Graur D, Pupko T. 2010. GUIDANCE: a web server for assessing alignment confidence scores. Nucleic Acids Res. 38:W23–W28.
- Prié V, Puillandre N. 2014. Molecular phylogeny, taxonomy, and distribution of French Unio species (Bivalvia, Unionidae). Hydrobiologia. 735:95–110.
- Robinson DF, Foulds LR. 1981. Comparison of phylogenetic trees. Math Biosci. 53:131–147.
- Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP, et al. 2012. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 61:539–542.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30:1312–1313.
- Whelan NV, Geneva AJ, Graf DL. 2011. Molecular phylogenetic analysis of tropical freshwater mussels (Mollusca: Bivalvia: Unionoida) resolves the position of Coelatura and supports a monophyletic Unionidae. Mol Phylogenet Evol. 61:504–514.
- Zouros E, Oberhauser Ball A, Saavedra C, Freeman KR. 1994. An unusual type of mitochondrial DNA inheritance in the blue mussel Mytilus. Proc Natl Acad Sci USA. 91:7463–7467.