
Abstract
In this study, we present the complete mitochondrial genome of the Bechstein’s bat, Myotis bechsteinii. The mitogenome is 17,151 bp in length and is AT-rich with base composition A (27.8%), C (22%), G (16.1%), and T (34.1%). The mitogenome shows conserved gene content and order similar with other mammalian mitogenomes, being composed of 13 protein-coding genes, 2 ribosomal RNA genes, 22 transfer RNA genes, and one control region. The majority of genes are encoded on the H-Strand except for ND6 and 8 tRNAs as found in other bat species. The field identification of Myotis bechsteinii was confirmed by phylogenetic analyses using datasets comprising whole mitogenomes and COXI. This mitogenome is a resource for future studies of Myotis bats and other mammals.
Keywords:
Myotis bechsteinii is a small, insectivorous bat species, typically found in forest areas. M. bechsteinii were wild caught in a forest near the city of Würzburg, Germany (latitude: N49°43′, longitude: E9°49′), where the species has been studied for more than two decades (Kerth & van Schaik Citation2012). A 3 mm diameter wing biopsy was taken and stored in silica beads prior to DNA extraction. DNA was extracted from an individual sample, DE6 (Teeling DNA Repository, UCD) using the Promega Wizard SV96 Genomic DNA purification system (catalogue no. A2371, Promega Corporation, Madison, WI). The entire mitogenome was amplified and sequenced using the primers, protocols, and sequencing procedures previously reported in Jebb et al. (Citation2015).
The entire mitogenome (GenBank accession no. KX757757) was assembled de novo using ABySS (v 1.5.2) (Simpson et al. Citation2009). The mitogenome was annotated using ARWEN (Laslett & Canbäck Citation2008) and Geneious (v 7.1.7) (Kearse et al. Citation2012), with M. myotis (Jebb et al. Citation2015) as a reference.
Following recommendations in Botero-Castro et al. (Citation2014), we phylogenetically validated the M. bechsteinii mitogenome. First, all Myotis COXI barcode sequences were downloaded from the Barcode Of Life Database (BOLD), and the assembled M. bechsteinii COXI sequence was added with a Murina ussuriensis COXI providing an outgroup. After quality control, 360 sequences were aligned with MAFFT (Katoh & Standley Citation2013) and resulted in a final alignment of 649 bp. Second, the M. bechsteinii whole mitogenome was aligned to 56 published, bat mitogenomes, and one outgroup (cow) using MAFFT, and the alignment was filtered using Gblocks (Castresana Citation2000). For both datasets, the GTR + Γ+I model of substitution was chosen under the AIC in jModeltest2 (Darriba et al. Citation2012). RAxML (v7.2.8) (Stamatakis Citation2006) was used to construct Maximum Likelihood (ML) phylogenies with 1000 bootstrap replicates to estimate support. The ML whole mitogenome phylogeny is shown in . MrBayes (v 3.2.6) (Huelsenbeck & Ronquist Citation2001) was used to construct a whole mitogenome phylogeny using Bayesian Inference (BI). Two iterations with 4 chains were run for 1.1 million generations and sampled every 200, with the first 100,000 generations discarded as burn-in. After 1.1 million generations, the analysis had reached stationarity and the iterations converged.
Figure 1. Whole mitogenome, Chiroptera phylogeny constructed using RAxML from an alignment of 57 Chiroptera mitogenomes, with cow (Bos taurus) as an outgroup. GenBank accession numbers are shown adjacent to species names. Percentage bootstrap support is shown at each node. Both ML and BI analyses show M. bechsteinii clusters within the genus Myotis. Nodes within Myotis which differed between the two analyses are marked with grey circles.
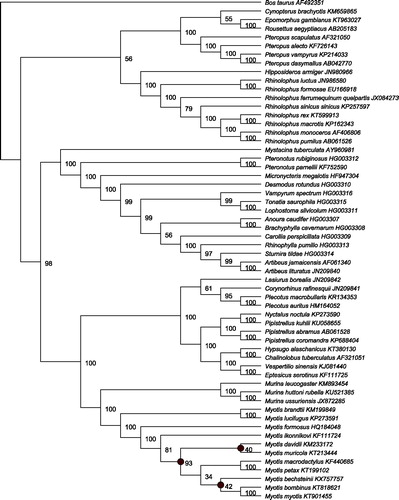
The assembled M. bechsteinii COXI sequence grouped exclusively with M. bechsteinii barcodes in the COXI only dataset, confirming species identity. In the whole mitogenome phylogeny, the M. bechsteinii mitogenome was placed within the Myotis genus, as shown in . The deep divergences within Myotis, amongst the Old World Clades, were poorly supported in both ML and BI analyses, and the exact relationships between the Eurasian clades differed between methods. These poorly supported deep nodes were previously reported by Ruedi et al. (Citation2013), and are unlikely to be resolved with a purely mitochondrial dataset such as this.
In this study, we sequenced, assembled, and phylogenetically validated the mitogenome of M. bechsteinii, a species whose population genetic structure and social behaviour has been intensively studied (Kerth & van Schaik Citation2012). This mitogenome is a resource for phylogenetic, biomedical, and on-going conservation studies.
Acknowledgements
We thank all the volunteers in Greifswald for their help in the field. We thank the European Research Council, who funded this work under grant number StG311000 awarded to Emma C. Teeling. Gerald Kerth’s work has been supported by the Deutsche Forschungsgemeinschaft DFG (KE 746/6-1).
Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
Additional information
Funding
References
- Botero-Castro F, Delsuc F, Douzery EJP. 2014. Thrice better than once: quality control guidelines to validate new mitogenomes. Mitochondrial DNA. 1736:1–6.
- Castresana J. 2000. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 17:540–552.
- Darriba D, Taboada GL, Doallo R, Posada D. 2012. jModelTest 2: more models, new heuristics and parallel computing. Nature Med. 9:772.
- Huelsenbeck JP, Ronquist F. 2001. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics (Oxford, England). 17:754–755.
- Jebb D, Foley NM, Puechmaille SJ, Teeling EC. 2015. The complete mitochondrial genome of the Greater Mouse-Eared bat, Myotis myotis (Chiroptera: Vespertilionidae). Mitochondrial DNA. 1736:1–3.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30:772–780.
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics (Oxford, England). 28:1647–1649.
- Kerth G, van Schaik J. 2012. Causes and consequences of living in closed societies: lessons from a long-term socio-genetic study on Bechstein?s bats. Mol Ecol. 21:633–646.
- Laslett D, Canbäck B. 2008. ARWEN: a program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics (Oxford, England). 24:172–175.
- Ruedi M, Stadelmann B, Gager Y, Douzery EJP, Francis CM, Lin L-K, Guillén-Servent A, Cibois A. 2013. Molecular phylogenetic reconstructions identify East Asia as the cradle for the evolution of the cosmopolitan genus Myotis (Mammalia, Chiroptera). Mol Biol Evol. 69:437–449.
- Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJM, Birol I. 2009. ABySS: a parallel assembler for short read sequence data. Genome Res. 19:1117–1123.
- Stamatakis A. 2006. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics (Oxford, England). 22:2688–2690.