
Abstract
The complete mitogenome of the Emerald Ash Borer (EAB, Agrilus planipennis) was obtained by gleaning mitochondrial sequences from whole-genome Illumina sequencing data. The circular genome has 15,942 base pairs and contains 13 protein-coding genes (PCGs), 22 transfer RNAs (tRNAs), 2 ribosomal RNAs (rRNAs) and an A–T-rich region. All PCGs begin with ATN codons. The nucleotide composition is highly asymmetric (31.65% A, 40.25% T, 17.39% G, 10.71% C), with an overall A–T content of 71.9%. Phylogenetic analysis based on insect mitogenomes indicated that EAB is closely related to other Buprestoidea species, clustering most closely with Chrysochroa fulgidissima.
The Emerald Ash Borer (EAB), Agrilus planipennis, is an invasive phloem-feeding insect pest of North American ash trees originating from East Asia (Herms & McCullough Citation2014). Since its initial discovery near the cities of Detroit (US) and Windsor (Canada) in 2002, EAB has spread rapidly to other US states and Canadian provinces. Genetic and genomic information are crucial elements to understand the movement of EAB in its introduced range. Likewise, full mitochondrial DNA information from EAB could help resolve the phylogeny of the genus Agrilus, which, with ca. 3000 species, is recognized as the most speciose genus in the animal kingdom (Jendek & Poláková Citation2014).
EABs were collected from infested ash trees in Toledo, Ohio (41°38'59.3"N 83°32'00.9"W). Genomic DNA was sequenced using the paired-end module on Illumina HiSeq2000. Five million pairs of raw reads were randomly chosen for reconstructing the mitogenome. Reads were filtered using Trimmomatic (LEADING: 20, TRAILING: 20, SLIDINGWINDOW: 4:15, Bolger et al. Citation2014). Quality-trimmed reads longer than 50 bp were used for the mitogenome construction with mitoMaker (http://sourceforge.net/projects/mitomaker/), using that of T. castaneum (GenBank accession number: KM009121) as a reference. The mitogenome was annotated using the MITOS web server (Bernt et al. Citation2013) and coding regions were manually verified by comparison against the NCBI nr database.
The mitogenome of EAB is 15,942 bp long (GenBank accession number: KT363854) and significantly biased towards A and T nucleotides (31.65% A, 40.25% T, 17.39% G, 10.71% C), similar to that of T. castaneum (A–T: 71.7%) (Friedrich & Muqim Citation2003). A total of 37 genes were identified, including 13 protein-coding genes (PCGs), 22 transfer RNAs (tRNAs), and 2 rRNAs. PCGs formed the largest portion (11,331 bp, 71.08%), while rRNAs and tRNAs accounted for 13.26% (2114 bp) and 9.12% (1454 bp), respectively. The A + T-rich region was 1184 bp long, which was located between the tRNA-Ile and 12S rRNA. All 13 PCGs were initiated by typical ATN codons (seven with ATG, four with ATT and two with ATA). Most PCGs ended with TAA codons except for ATP8 and CYTB, which had TAG codons.
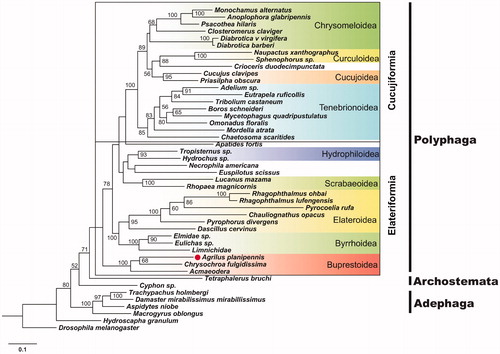
Maximum likelihood (ML) analyses were performed to investigate EAB phylogenetic relationships with other coleopterans using the 44 mitogenome sequences available for this order. The 13 PCGs were concatenated and aligned with MUSCLE (Edgar Citation2004) using default parameters. The ML analysis was carried out using RAxML (Stamatakis Citation2014), with the optimal substitution model of MeREV, which was selected by Protest3 under the Akaike Information Criterion (AIC). As shown in , the phylogenetic analysis placed EAB within the Buprestoidea, and its closest relative is Chrysochroa fulgidissima. This complete mitogenome sequence will provide a resource for EAB population genetic research, and may help map routes of EAB invasions.
Figure 1. Maximum likelihood estimation of the phylogenetic relationships among EAB and other species within insect order of Coleoptera. Bootstrap values were calculated using 100 bootstrap pseudoreplicates; only values >50 are shown.
Acknowledgements
We thank Drs Rick Westerman and Philip San Miguel (Purdue University Genomics Core Facility) for Illumina sequencing services.
Disclosure statement
The authors report no conflicts of interest. The authors are responsible for the content and writing of the paper. This work was funded by a Genomics Research and Development Initiative (GRDI) grant from NRCan to DD, GQ and MC.
Additional information
Funding
References
- Bernt M, Donath A, Juhling F, Externbrink F, Florentz C, Fritzsch G, Putz J, Middendorf M, Stadler PF. 2013. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 69:313–319.
- Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30:2114–2120.
- Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32:1792–1797.
- Friedrich M, Muqim N. 2003. Sequence and phylogenetic analysis of the complete mitochondrial genome of the flour beetle Tribolium castanaeum. Mol Phylogenet Evol. 26:502–512.
- Herms DA, McCullough DG. 2014. Emerald ash borer invasion of North America: history, biology, ecology, impacts, and management. Annu Rev Entomol. 59:13–30.
- Jendek E, Poláková J. 2014. Host plants of world Agrilus (Coleoptera, Buprestidae): a critical review. Cham, Switzerland: Springer International Publishing AG.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30:1312–1313.