
Abstract
Here, we describe the first mitochondrial genome of the angelshark, Squatina squatina. The genome is 16,689 bp in length with 13 protein-coding genes, 22 tRNA genes, 2 rRNA genes, and a non-coding control region. Base composition of the mitogenome has an A + T bias (62.9%), seen commonly in other elasmobranchs. This genome provides a key resource for future investigations of the population genetic dynamics and evolution of this Critically Endangered shark.
The angelshark, Squatina squatina (Squatinidae), once ranged throughout the Northeast Atlantic, Mediterranean, and Black Sea. With a benthic lifestyle and low productivity, S. squatina is particularly Vulnerable to overexploitation, especially by demersal trawl fisheries (Miller Citation2016). Given estimates of over 80% depletion of its historical abundance, this species is designated as Critically Endangered on the International Union for the Conservation of Nature (IUCN) Red List (Ferretti et al. Citation2015). Despite its status as a species of high conservation concern, minimal genetic assessment of the angelshark has been performed to help inform conservation management.
Individual mitochondrial gene sequences are widely used to assess species diversity and population connectivity, however, whole mitogenome analysis can help identify regions of high variability, and provide higher resolution of intra-species diversity, connectivity, divergence time estimates, and phylogenies (Feutry et al. Citation2014). Here, we report the first mitochondrial genome sequence of the angelshark, S. squatina. The individual sequenced was caught as bycatch off the north east coast of Gran Canaria in the Canary Islands in 2008 as part of the islands’ artisanal fishery (Osaer et al. Citation2015). The sample (accession number OC-257) is stored in 100% ethanol at Nova Southeastern University, College of Natural Sciences and Oceanography.
Five overlapping sections of the S. squatina mitogenome were amplified by long PCR using five primer pairs designed from the consensus sequences of three published Squatinidae mitogenomes (S. formosa NC025328; S. japonica NC024276; S. nebulosa NC025578) with Geneious v.7.0.6 (http://geneious.com, Kearse et al. Citation2012) and its built-in Primer3 Design Tool (Untergasser et al. Citation2012). The PCR amplicons were pooled for library preparation with a Nextera XT DNA Sample Preparation kit (Illumina, San Diego, CA). Final whole mitogenome libraries were 2 × 250 bp paired-end sequenced, on an Illumina MiSeq sequencer. Reads were de novo assembled with CLC Genomics Workbench (QIAGEN) as well as aligned with reference-based assembler Bowtie2 (Langmead & Salzberg Citation2012) to resolve ambiguities. The mitogenome was annotated using MitoAnnotator (Iwasaki et al. Citation2013) and annotations confirmed by comparison to the three other published Squatina species.
Figure 1. Bayesian tree depicting currently available Squatinidae mitogenomes with closely related outgroup. Scale and clade posterior probabilities are displayed. Labels include species name and GenBank RefSeq accession numbers.
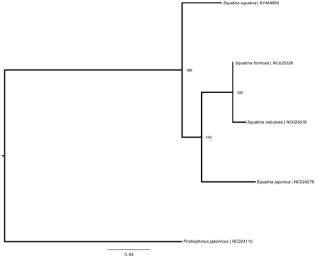
The S. squatina mitogenome sequence (gb: KY464954) is 16,689 bp in length with a gene order typical of most vertebrates, with 13 protein-coding genes, 22 tRNA genes, 2 rRNA genes, and a non-coding control region (D-loop). Nucleotide composition leaned to an A + T bias with 30.9% A, 23.4% C, 13.7% G, and 32.0% T. The program MUSCLE was used to align the available squatinid mitogenome sequences on NCBI, with Pristiophorus japonicus (gb: NC024110) as an outgroup. A Bayesian phylogeny was constructed in MrBayes 3.2 (Huelsenbeck & Ronquist Citation2001; Ronquist & Huelsenbeck Citation2003) using the GTR + I as the best substitution model given by jModelTest v.2.1.10 (Darriba et al. Citation2012) Bayesian Information Criterion. The tree () shows the Atlantic restricted S. squatina as a sister species to the other three squatinids of Pacific origin. This first S. squatina mitogenome provides a genomic resource to aid conservation and management efforts for this highly depleted species.
Acknowledgements
We thank Dr Jose Lopez and Jorie Skutas of the Nova Southeastern University Microbiology and Genetics Lab for use of their Illumina MiSeq.
Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
Additional information
Funding
References
- Darriba D, Taboada GL, Doallo R, Posada D. 2012. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods. 9:772.
- Ferretti F, Morey G, Serena F, Mancusi C, Fowler SL, Dipper F, Ellis J. 2015. Squatina squatina, Angel shark. The IUCN Red List of Threatened Species 2015:e.T39332A48933059. Available from: http://dx.doi.org/10.2305/IUCN.UK.2015-1.RLTS.T39332A48933059.en
- Feutry P, Kyne PM, Pillans RD, Chen X, Naylor GJP, Grewe PM. 2014. Mitogenomics of the Speartooth Shark challenges ten years of control region sequencing. BMC Evol Biol. 14:232
- Huelsenbeck JP, Ronquist F. 2001. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 17:754–755.
- Iwasaki W, Fukunaga T, Isagozawa R, Yamada K, Maeda Y, Satoh TP, Sado T, Mabuchi K, Takeshima H, Miya M, Nishida M. 2013. Mitofish and mitoannotator: a mitochondrial genome database of fish with an accurate and automatic annotation pipeline. Mol Biol Evol. 30:2531–2540.
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28:1647–1649.
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nature Meth. 9:357–359.
- Miller MH. 2016. Status Review Report of 3 Species of Angelsharks: Squatina aculeata, S. oculata, and S. squatina. Report to National Marine Fisheries Service, Office of Protected Resources. June 2016. p. 74.
- Osaer F, Narváez K, Paiuelo JG, Lorenzo JM. 2015. Sexual development and maturity scale for the angelshark Squatina squatina (Elasmobranchii: Squatinisae), with comments on the adequacy of general maturity scales. Sex Early Dev Aquat Org. 1:117–132.
- Ronquist F, Huelsenbeck JP. 2003. MRBAYES 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 19:1572–1574.
- Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, Rozen SG. 2012. Primer3 - new capabilities and interfaces. Nucleic Acids Res. 15:e115.