
Abstract
Here we present the first complete mitochondrial genome of the lesser sac-winged bat Saccopteryx leptura (Chiroptera: Emballonuridae) from Costa Rica, assembled from next-generation sequencing data. The mitogenome of Saccopteryx leptura measures 16,577 bp in length, and contains 13 protein-coding genes, 2 ribosomal RNA genes and 22 transfer RNA genes. A slight A + T bias was observed in the mitogenome of Saccopteryx leptura with an overall base composition of 31.5% A, 28.3% T, 25.8% C, and 14.2% G, and a GC content of 40.1%. The gene arrangement was identical to that of previously described bat mitogenomes.
The family of Emballonuridae comprises 51 bat species and is found in the old as well as in the new world (Simmons Citation2005). The Neotropical genera and especially the genus Saccopteryx have recently received much attention since several species show complex social behaviour including unusual mammalian sex-biased dispersal (Nagy et al. Citation2007) and vocal learning (Knörnschild Citation2014). Finally, high genetic divergence in the mitochondrial genome was found throughout the species’ distribution ranges indicating the existence of cryptic species diversity (Clare et al. Citation2011). Reliable phylogenetic and population genetic analyses involving also the mitochondrial genome will help to understand the divergent evolution of behavioural traits within the Neotropical emballonurids. Here we present the full mitogenome of the Neotropical bat species Saccopteryx leptura (Schreber 1774).
An adult male was caught in Costa Rica at La Selva Biological Station (10.4311°N, 84.0052°W) on 21 August 2008 (research permit 183-2008-SINAC). A small wing tissue biopsy was taken prior to the release of the bat.
Genomic DNA was isolated according to a salt-chloroform procedure (modified from Müllenbach et al. Citation1989). Five hundred nanograms were used for fragmentation with Covaris Ultrasonificator (AFA Technology, Woburn, MA). Hundred nanograms of sheared DNA were used for Illumina Library Preparation with NEXTflex Rapid DNASeq Kit (Biooscientific, Austin, TX). 150 bp Paired End Sequencing was performed with Illumina´s HiSeq 2500 Sequencer (Rapid Mode, San Diego, CA) using one third of a flowcell. Shotgun sequencing yielded a total of 70,558,564 paired end reads. A complete circularized mitochondrial genome was obtained with NOVOplasty 2.4 (Dierckxsens et al. Citation2017) using kmer 47. Annotations were carried out with MITOchondrial genome annotation Server (MITOS) (Bernt et al. Citation2013), and manual validation of the coding regions was done using the NCBI ORF Finder (http://www.ncbi.nlm.nih.gov/gorf/gorf.html).
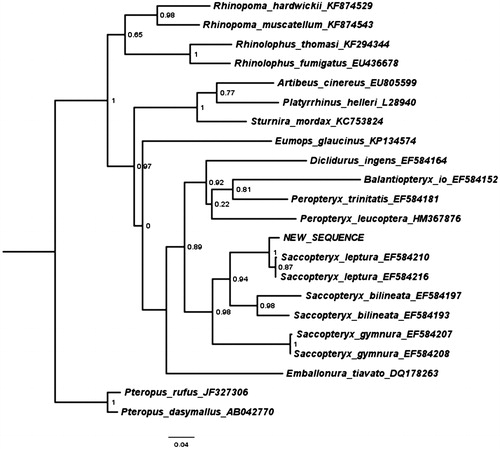
The annotated sequence file was submitted to NCBI (accession no. KY681816). The phylogenetic position of the new sequence of S. leptura based on the cytochrome B gene is shown in .
Figure 1. Maximum likelihood tree illustrating the phylogenetic position of the newly sequenced Saccopteryx leptura gene sequence among a subset of bat species. Cytochrome B sequences were aligned using MAFFT 7.271 and highly divergent or poorly aligned regions were removed with Gblocks 0.91b (Castresana Citation2000) allowing for gap positions and smaller blocks. Trees were calculated using PhyML 3.1 (Guindon et al. Citation2010) with four rate categories, optimized equilibrium frequencies, GTR model of sequence evolution and combined heuristics (Nearest Neighbour Interchange and Subtree Pruning and Rerafting).
The complete mitochondrial transcript of Saccopteryx leptura was 16,577 bp in length and contained 13 protein-coding genes (PCGs), 2 ribosomal RNA genes and 22 transfer RNA genes. As described for other bat mitogenomes (Yoon & Park Citation2016; Yoon et al. Citation2016), the mitochondrial genome of Saccopteryx leptura contained a slight A + T bias with an overall base composition of 31.5% A, 28.3% T, 25.8% C, and 14.2% G, and a GC content of 40.1%. The gene arrangement of the present mitogenome is similar to that of other bats (Yu et al. Citation2016; Jiang et al. Citation2016). Most of the genes were encoded on the L-strand except for ND6 and eight tRNA genes (tRNAGln, tRNAAla, tRNAAsn, tRNACys, tRNATyr, tRNASer2, tRNAGlu and tRNAPr°), which were encoded in the H-strand. All PCGs had ATG as initiation codon with the exception of ND3 and ND5 which used ATA as initiation codon. TAA was the most used termination codon except for CYTB, which used an AGA termination codon. Incomplete stop codons (T– or TA–) were found in COX3 and ND4. The 12S and 16S genes had a length of 967 and 1559 bp, respectively.
Disclosure statement
The authors declare no conflict of interest.
References
- Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G, Pütz J, Middendorf M, Stadler PF. 2013. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 69:313–319.
- Castresana J. 2000. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 17:540–552.
- Clare EL, Lim BK, Fenton MB, Hebert PDN. 2011. Neotropical bats: estimating species diversity with DNA barcodes. PLoS One. 6:e22648
- Dierckxsens N, Mardulyn P, Smits G. 2017. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucl Acids Res. 45:e18–e18.
- Guindon S, Dufayard J-F, Lefort V, Anisimova M, Hordijk W, Gascuel O. 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 59:307–321.
- Jiang J-J, Wang S-Q, Li Y-J, Zhang W, Yin A-G, Hu M. 2016. The complete mitochondrial genome of insect-eating Brandt’s bat, Myotis brandtii (Myotis, Vespertilionidae). Mitochondrial DNA Part A. 27:1403–1404.
- Knörnschild M. 2014. Vocal production learning in bats. Curr Opin Neurobiol. 28:80–85.
- Müllenbach R, Lagoda PJL, Welter C. 1989. An efficient salt-chloroform extraction of DNA from blood and tissues. Trends Genet. 5:391
- Nagy M, Heckel G, Voigt CC, Mayer F. 2007. Female-biased dispersal and patrilocal kin groups in a mammal with resource-defence polygyny. Proc Biol Sci. 274:3019–3025.
- Simmons NB. 2005. Mammalian species of the world: a taxonomic and geographic reference. Chapter Order Chiroptera. Baltimore (MD): Johns Hopkins University Press. p. 312–529.
- Yoon GB, Park YC. 2016. The complete mitogenome of the Korean greater tube-nosed bat, Murina leucogaster (Chiroptera: Vespertilionidae). Mitochondrial DNA Part A. 27:2145–2146.
- Yoon KB, Lee JH, Cho JY, Park YC. 2016. The complete mitochondrial genome of the Asian particolored bat Vespertilio sinensis (Chiroptera: Vespertilionidae) in Korea. Mitochondrial DNA Part A. 27:299–300.
- Yu D, Qian K, Storey KB, Hu Y, Zhang J. 2016. The complete mitochondrial genome of Myotis lucifugus (Chiroptera: Vespertilionidae). Mitochondrial DNA Part A. 27:2423–2424.