
Abstract
The bat-eared fox, Otocyon megalotis, is the only member of its genus and is thought to occupy a basal position within the dog family. These factors can lead to challenges in complete mitochondrial reconstructions and accurate phylogenetic positioning. Here, we present the first complete mitochondrial genome of the bat-eared fox recovered using shotgun sequencing and iterative mapping to three distantly related species. Phylogenetic analyses placed the bat-eared fox basal in the Canidae family within the clade including true foxes (Vulpes) and the raccoon dog (Nyctereutes) with high support values. This position is in good agreement with previously published results based on short fragments of mitochondrial and nuclear genes, therefore adding more support to the basal positioning of the bat-eared fox within Canidae.
The bat-eared fox (Otocyon megalotis) is a small member of the Canidae family and the only species of the genus Otocyon. It occurs in two allopatric populations across Africa (Clark Citation2005) and is considered a basal canid species (Sillero-Zubiri & Macdonald Citation2004). Studies using short mitochondrial and nuclear genes support a basal placement of the bat-eared fox within Canidae, as sister group to the clade including true foxes (Vulpes) and the Raccoon dog (Nyctereutes procyonoides) (Lindblad-Toh et al. Citation2005). However, despite previous genetics studies, the complete mitochondrial genome of the bat-eared fox has so far not been published.
Our bat-eared fox sample was captured on Benfontein game farm outside of Kimberley, central South Africa (28°99′ S, 24°81′ E, e.g. le Roux et al. (Citation2014)) under permits from the animal care and use committee of the University of Pretoria (EC031-07) and from the provincial government in the Northern Cape (FAUNA 846/2009, FAUNA 847/2009).
DNA was extracted using a Zymo genomic DNA clean and concentrator extraction kit, built into Nextera Illumina sequencing libraries and sequenced on an Illumina Nextseq 500 (University of Potsdam, Germany). We trimmed raw reads using Cutadapt v1.4 (Martin Citation2011), merged overlapping fragments using FLASH v1.2.10 (Magoč & Salzberg Citation2011), and removed duplicate reads using Prinseq (Schmieder & Edwards Citation2011).
We undertook iterative mapping using MITObim v1.8 (Hahn et al. Citation2013) with default parameters apart from the mismatch value, which was 3%. Three independent runs were performed using different bait sequences from three species; Canis lupus (Genbank accession KC461238.1), Vulpes vulpes (Genbank accession JN711443.1), and Urocyon littoralis (Genbank accession KP128962.1). Consensus sequences were called using a minimum read coverage of 10× and a 75% base call threshold in Geneious v9.0.5 (Kearse et al. Citation2012). Automatic annotation was performed using MITOS (Bernt et al. Citation2013).
We aligned our sequence with representatives of Carnivora using Mafftv7.271 (Katoh & Standley Citation2013). We constructed a phylogenetic tree using BEAST version 1.8.4 (Drummond et al. Citation2012) on the Cipres server (Miller et al. Citation2010) specifying GTR I + G as the substitution model, as determined using Jmodeltest v2.1.7 (Posada Citation2008) and a Yule speciation model (Yule Citation1925; Gernhard Citation2008).
Our bat-eared fox mitochondrial sequence (Genbank accession KY776502) contains 16,431 bp. The genomes produced were identical regardless of starting bait reference. Our bat-eared fox mitochondrial assembly contained all 13 protein-coding genes with expected open reading frames, 22 transfer RNA genes, and 2 ribosomal RNA genes found within a typical vertebrate mitochondrial genome.
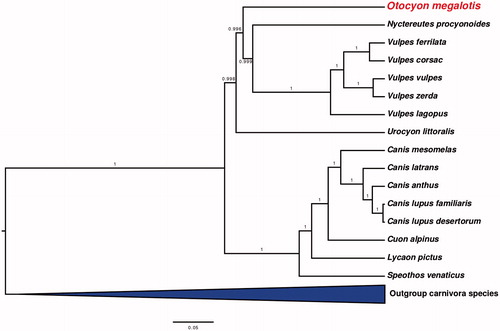
Phylogenetic analyses found further support for a basal placement of the bat-eared fox within the Canidae. Otocyon was placed with high confidence (posterior probabilities > 0.99) as sister to the clade containing both the raccoon dog (Nyctereutes) and true foxes (Vulpes) (). This result is consistent with previous conclusions based on short regions of mitochondrial and nuclear genes (Lindblad-Toh et al. Citation2005).
Figure 1. Bayesian tree showing the phylogenetic positioning of Otocyon megalotis within the Canidae and other clades within Carnivora. Numbers on branches represent posterior probabilities.
Acknowledgements
We would like to thank Armanda Bastos for the use of her laboratory facilities at the University of Pretoria, South Africa.
Disclosure statement
We would like to declare no conflicts of interest.
Additional information
Funding
Related Research Data
References
- Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G, Pütz J, Middendorf M, Stadler PF. 2013. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 69:313–319.
- Clark HO Jr. 2005. Otocyon megalotis. Mamm Species. 76:1–5.
- Drummond AJ, Suchard MA, Xie D, Rambaut A. 2012. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 29:1969–1973.
- Gernhard T. 2008. The conditioned reconstructed process. J Theor Biol. 253:769–778.
- Hahn C, Bachmann L, Chevreux B. 2013. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads—a baiting and iterative mapping approach. Nucleic Acids Res. 41:e129.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30:772–780.
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28:1647–1649.
- le Roux A, Beishuizen R, Brekelmans W, Ganswindt A, Paris M, Dalerum F. 2014. Innovative parental care in a myrmecophageous mammal. Acta Ethol. 17:63–66.
- Lindblad-Toh K, Wade CM, Mikkelsen TS, Karlsson EK, Jaffe DB, Kamal M, Clamp M, Chang JL, Kulbokas EJ, III Zody MC, et al. 2005. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature. 438:803–819.
- Magoč T, Salzberg SL. 2011. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 27:2957–2963.
- Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.j. 17:10–12.
- Miller MA, Pfeiffer W, Schwartz T. 2010. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In: 2010 Gateway Computing Environments Workshop (GCE). p. 1–8.
- Posada D. 2008. jModelTest: phylogenetic model averaging. Mol Biol Evol. 25:1253–1256.
- Schmieder R, Edwards R. 2011. Quality control and preprocessing of metagenomic datasets. Bioinformatics. 27:863–864.
- Sillero-Zubiri C, Macdonald DW. 2004. The biology and conservation of wild canids. Oxford: Oxford University Press.
- Yule GU. 1925. A mathematical theory of evolution, based on the conclusions of Dr. JC Willis, FRS. Proc Math Phys Eng Sci. 213:21–87.