
Abstract
Heritiera angustata (Malvaceae) is an evergreen tree distributed in the Chinese provinces of Hainan and Yunnan and in Cambodia. In China, it is listed as ‘Endangered’ (EN) China Red List of Higher Plants, although it is not protected by law. The complete chloroplast (cp) genome sequence of this threatened species is reported in this study, based on high-throughput sequencing (Illumina). The complete cp genome is 168, 953 bp in length, containing a pair of inverted repeat regions (IRs) of 34,491 bp, a large single copy (LSC) region of 89,054 bp, and a small single copy (SSC) region of 10,917 bp. The cp genome contains 129 genes, consisting of 85 protein-coding genes, 36 tRNA genes and eight rRNA genes. The overall AT content in the cp genome of H. angustata is 63.2%. The phylogenetic analyses indicate that there is a close relationship between H. angustata and Firmiana pulcherrima.
Heritiera angustata Pierre (Malvaceae) is an evergreen tree widely distributed in the mountains or near coastal areas in southeast region of Hainan and in Yunnan provinces of China, as well as in Cambodia (Tang et al. Citation2007). It is listed as ‘Endangered’ (EN) in the China Red List of Higher Plants (MEP-CAS Citation2013). Despite its threatened status, it is not protected by law in China, although some of its populations are included within nature reserves. Genetic diversity is pre-requisite for evolutionary adaptation; thus, preservation of genetic diversity should be a key factor in species’ conservation plans (Hamrick et al. Citation1991). In order to inform a comprehensive conservation strategy for this threatened species, an improved understanding of its genomics information is urgently needed. Here, the complete chloroplast genome of H. angustata (GenBank: this study) is determined and presented, based on the Illumina paired-end sequencing data.
In this study, we sampled a healthy individual of H. angustata from Diaoluo Mountain (18.67°N, 109.88°E), which is a National Nature Reserve of Hainan, China. We employed the modified CTAB method (Doyle and Doyle Citation1987) to extract the total genomic DNA of H. angustata from silica gel-dried leaves. A voucher specimen of H. angustata (H.-F. Wang B38) was deposited at the Institute of Tropical Agriculture and Forestry, Hainan University (Haikou, China). Whole-genome short-gun sequencing was performed on the Illumina Hiseq 2500 platform, with the 150 bp paired-end sequencing method. After filtering and trimming with NGSQC-Toolkit v2.3.3 (Patel and Jain Citation2012), the clean reads with an average coverage of 300 × were used to conduct the referenced-based chloroplast genome assembly with the program MITObim v1.8 (Hahn et al. Citation2013). All genes were annotated with the program of GENEIOUS R8.0.2 (Biomatters Ltd., Auckland, New Zealand). The chloroplast genome of Theobroma cacao (HQ336404.2) (Jansen et al. Citation2011) was used as the reference for assembling and annotation. The annotation was corrected with Dual Organellar Genome Annotator (DOGMA) software (Wyman et al. Citation2004), and a circular plastid genome map was generated with OGDRAW (Lohse et al. Citation2013).
The cpDNA of H. angustata was a circular molecule of 168,953 bp with a quadripartite structure, containing a pair of inverted repeats (IRs) of 34,491 bp, separated by a large single copy (LSC) region, and a small single copy (SSC) region of 89,054 and 10,917 bp, respectively. It contained 129 genes, including 85 protein-coding genes (76 PCG species), eight ribosomal RNA genes (four rRNA species) and 36 tRNA genes (29 tRNA species). Among these, 13 genes (trnA-UGC, trnI-GAU, trnK-UUU, trnL-UAA, trnV-UAC, atpF, ndhA, ndhB, petB, petD, rpoC1, rpl2, and rpl16) harboured a single intron and one gene (ycf3) had two introns. The majority of gene species in H. angustata occurred as a single copy. The overall AT content of H. angustata chloroplast genome was 63.2%, while the corresponding values of the LSC, SSC and IR regions were 65.1%, 68.28% and 59.95%, respectively.
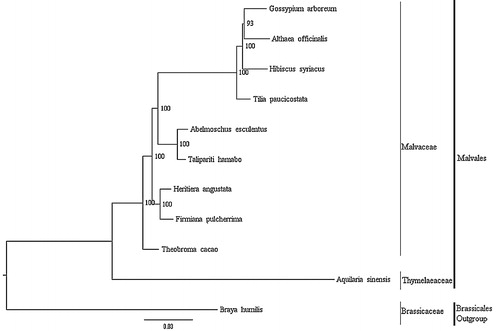
A maximum likelihood (ML) phylogenetic tree of the nine published complete chloroplast genomes of Malvales (plus H. angustata) was built with RAxML (Stamatakis Citation2006), using Braya humilis (Brassicaceae, Brassicales) as outgroup (). The phylogenetic analysis indicated that all members of Malvales were clustered with a high bootstrap support (BS) value and there was a close relationship between H. angustata and Firmiana pulcherrima. In this paper, we report the characterization of the complete chloroplast genome of H. angustata for the first time, which may provide a useful resource for conservation genetics studies of H. angustata, and also for phylogenetic studies of Malvales.
Figure 1. Phylogenetic tree based on 10 complete chloroplast genome sequences of Malvales. Accession numbers:, Abelmoschus esculentus NC_035234.1, Althaea officinalis NC_034701.1, Aquilaria sinensis KT148967.1, Firmana pulcherrima MF621982.1, Gossypium arboreum HQ325740.1, H. angustata (this study), Hibiscus syriacus KR259989.1, Talipariti hamabo NC_030195.1, Theobroma cacao HQ336404.2, Tilia paucicostata NC_028591.1; outgroup: Braya humilis KY912032.1.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Doyle JJ, Doyle JL. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 19:11–15.
- Hahn C, Bachmann L, Chevreux B. 2013. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads-a baiting and iterative mapping approach. Nucleic Acids Res. 41:e129.
- Hamrick JL, Godt MJW, Murawski DA, Loveless MD. 1991. Correlations between species traits and allozyme diversity: implications for conservation biology. In: Falk DA, Holsinger KE, editors. Genetics and conservation of rare plants. New York: Oxford University Press; p. 75–86.
- Jansen RK, Saski C, Lee S-B, Hansen AK, Daniell H. 2011. Complete plastid genome sequences of three rosids (Castanea, Prunus, Theobroma): evidence for at least two independent transfers of rpl22 to the nucleus. Mol Biol Evol. 28:835–847.
- Lohse M, Drechsel O, Kahlau S, Bock R. 2013. OrganellarGenome-DRAW: a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 41:W575–W581.
- Ministry of Environmental Protection–Chinese Academy of Sciences (MEP-CAS). 2013. China red list of higher plants: evaluation’s report. Ministry of Environmental Protection of the People’s Republic of China & Chinese Academy of Sciences, Beijing [in Chinese].
- Patel RK, Jain M. 2012. NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PLoS One. 7:e30619.
- Stamatakis A. 2006. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 22:2688–2690.
- Tang Y, Gilbert MG, Dorr LJ. 2007. Sterculiaceae. In: Wu ZY, Raven PH, Hong DY, editors. Flora of China. Vol. 12 (Hippocastanaceae through Theaceae). Beijing, St. Louis: Science Press and Missouri Botanical Garden Press; p. 302–330.
- Wyman SK, Jansen RK, Boore JL. 2004. Automatic annotation of organellar genomes with DOGMA. Bioinformatics. 20:3252–3255.