
Abstract
Nomocharis pardanthina Franchet (Liliaceae) is an endangered species naturally distributed in China. The complete chloroplast genome sequence of N. pardanthina was generated by de novo assembly using whole genome next generation sequencing data. The chloroplast genome of N. pardanthina was 152,718 bp in length and divided into four distinct regions, such as large single copy region (82,154 bp), small single copy region (17,524 bp) and a pair of inverted repeat regions (26,520 bp). The genome annotation predicted a total of 112 genes, including 78 protein-coding genes, 30 tRNA genes, and 4 rRNA genes. Phylogenetic analysis with the reported chloroplast genomes revealed that N. pardanthina is nested in Lilium and has close relationship to L. bakerianum and L. taliense.
Keywords:
Nomocharis pardanthina Franchet (Liliaceae) is an herbaceous plant species naturally distributed in China. It wildly grows in forest margins and grassy slopes at an elevation of 2700–4100 m (Liang and Tamura Citation2000). Nomocharis pardanthina shows unique flower morphology different from other species in Liliaceae and is placed in the genus Nomocharis Franchet. However, recent molecular phylogenetic analysis indicates that Nomocharis is nested within Lilium (Hayashi and Kawano Citation2000; Rønsted et al. Citation2005; Gao et al. Citation2012, Citation2013). Although several phylogenetic studies reported nuclear and chloroplast sequences of N. pardanthina, the complete chloroplast genome sequence is not available till now. Here, we report the complete chloroplast genome sequence of N. pardanthina to provide a genomic resource and to clarify phylogenetic relationship of this plant with other species in the Liliaceae family.
Total genomic DNA was isolated from mature leaves sampled from Mt. Gaoligong (27°47′23″N 98°27′04″E), Yunnan Province, China. Voucher specimens were deposited in SZ (Sichuan University Herbarium). Total genomic DNA was extracted by Plant Genomic DNA Kit. The isolated genomic was manufactured to average 350 bp paired-end(PE) library using Illumina Hiseq platform (Illumina, San Diego, CA), and sequenced by Illumina genome analyser (Hiseq PE150). Owing to the inference of nuclear genome, chloroplast genome-related reads were sieved by mapping to the closer species L. taliense. Contigs, assembled using SOAPdenovo2 (Luo et al. Citation2012), were sorted and joined into a single-draft sequence using Geneious (Kearse et al. Citation2012), by comparison with the chloroplast sequence of L. taliense as a reference. Gapcloser was used to fill the gapped sites, and the draft sequence was corrected manually by clean read mapping using bowtie2 (Langmead and Salzberg Citation2012) and Tablet (Milne et al. Citation2013). The genes in chloroplast genome were predicted using Geneious and corrected manually.
The complete chloroplast genome of N. pardanthina (GenBank accession no. MG704135) was a circular form of 152,718 bp in length, which was separated into four distinct regions such as large single copy (LSC) region of 82,154 bp, small single copy (SSC) region of 17,524 bp and a pair of inverted repeat regions of 26,520 bp. Overall GC contents of chloroplast genomes were 37.00%. The chloroplast genome contained a total of 112 genes including 78 protein-coding genes, 30 tRNA genes, and 4 rRNA genes.
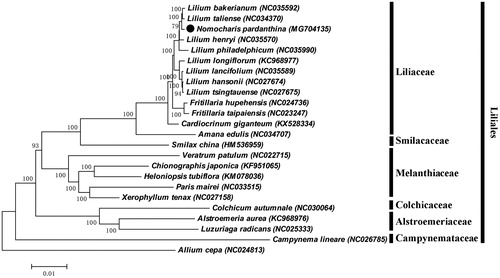
In order to understand the phylogenetic relationship between N. pardanthina and related species, the complete chloroplast genome sequence of 16 genera (24 species) were aligned by MAFFT (Katoh et al. Citation2002) and trimmed by trimAl (Capella-Gutierrez et al. Citation2009). The evolution history was inferred by using maximum-likelihood method based on Tamura-Nei model in MAGA7.0 (Kumar et al. Citation2016). Bootstrap (BS) value was calculated from 1000 replicate analysis. The phylogenetic tree was divided into seven independent groups, which were Amaryllidaceae, Campynemataceae, Alstroemeriaceae, Colchicaceae, Melanthiaceae, Smilacaceae, and Liliaceae (). As was expected, N. pardanthina was closely grouped with the genus Lilium, and occurred as a sister group with clade of L. bakerianum and L. taliense with 100% BS value.
Figure 1. ML phylogenetic tree of N. pardanthina with 22 species in the Liliales order was constructed by chloroplast genome sequences. Numbers on the nodes are bootstrap values from 1000 replicates. Allium cepa was selected as an outgroup.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Capella-Gutierrez S, Silla-Martinez JM, Gabaldon T. 2009. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 25:1972–1973.
- Gao YD, Hohenegger M, Harris AJ, Zhou SD, He XJ, Wan J. 2012. A new species in the genus Nomocharis Franchet (Liliaceae): evidence that brings the genus Nomocharis into Lilium. Plant Syst E. 298:69–85.
- Gao YD, Harris AJ, Zhou SD, He XJ. 2013. Evolutionary events in Lilium (including Nomocharis, Liliaceae) are temporally correlated with orogenies of the Q–T plateau and the Hengduan Mountains. Mol Phylogenet Evol. 68:443–460.
- Hayashi K, Kawano S. 2000. Molecular systematics of Lilium and allied genera (Liliaceae): phylogenetic relationships among Lilium and related genera based on the rbcL and matK gene sequence data. Plant Species Biol. 15:73–93.
- Katoh K, Misawa K, Kuma K, Miyata T. 2002. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30:3059–3066.
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28:1647–1649.
- Kumar S, Stecher G, Tamura K. 2016. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 33:1870–1874.
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods. 9:357–U354.
- Liang SY, Tamura M. 2000. Nomocharis. In: Wu ZY, Raven PH, editors. Flora of China. Vol. 24. St. Louis: Science Press/Beijing: Missouri Botanical Garden Press; p. 149–150.
- Luo RB, Liu BH, Xie YL, Li ZY, Huang WH, Yuan JY, He GZ, Chen YX, Pan Q, Liu YJ, et al. 2012. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience. 1:18.
- Milne I, Stephen G, Bayer M, Cock PJA, Pritchard L, Cardle L, Shaw PD, Marshall D. 2013. Using Tablet for visual exploration of second-generation sequencing data. Brief Bioinform. 14:193–202.
- Rønsted N, Law S, Thornton H, Fay MF, Chase MW. 2005. Molecular phylogenetic evidence for the monophyly of Fritillaria and Lilium (Liliaceae; Liliales) and the infrageneric classification of Fritillaria. Mol Phylogenet Evol. 35:509–527.