
Abstract
Taiwan Blue Magpie (Urocissa caerulea) is endemic to Taiwan and listed as threatened species protected by law. In this study, we first determined and described the complete mitochondrial genome of Taiwan Blue Magpie. The circle genome is 16,928 bp in length, and contains 13 protein coding, 22 tRNA, two rRNA genes, and one non-coding control region (CR). The overall base composition of the mitochondrial DNA is 30.99% for A, 24.69% for T, 30.07% for C, and 14.25% for G. The percentage of G + C content is 44.32%. This work provides fundamental molecular data which will be useful for evolution and phylogeny studies on Corvidae in the future.
Urocissa is a genus of birds in the family Corvidae, which includes six species distributed in Indian subcontinent, Indochina, Southern China, and Taiwan (BirdLife International Citation2016; Integrated Taxonomic Information System Citation2018). Though members of genus Urocissa have a wide distribution range and are appealing for their appearances, little phylogeny research information such as sequence data are available at present. Here, a complete mitochondrial genome of Taiwan Blue Magpie is presented. The Taiwan Blue Magpie (Urocissa caerulea) is an endemic species in Taiwan, which lives in broadleaf forests or parks with abundant trees from lowland to mid-elevation. The material we used in this study was the health-examination blood sample kept in Wildlife Cryobank, Taipei Zoo (deposit no. 20170210D10). The voucher of blood sample is D1818, a male Taiwan Blue Magpie transported from Hsinchu Zoo on 3 July 2017, and is kept in Taipei Zoo currently. The usage of this blood sample was authorized by Forestry Bureau, Council of Agriculture, Executive Yuan, Taiwan, with the permission document number 1061700466 (2017-19). Genomic DNA was extracted using the DNeasy Blood & Tissue Kit (QIAGEN, Hilden, Germany). The PCR and sequencing experiments were carried out mainly by the following procedures described by Chiang et al. (Citation2017). We designed two pairs of long PCR primers in the beginning (TBM-1F: 5′-CATCATCTGAGAGGCTTTCGCATCC3-3′ and TBM-1R: 5′-CTCAGGCTCATTCTACTAGTGTTTGTC-3′; TBM-2F: 5′-AACAAAGAGACCTGAAACATCGGAGTA-3′, and TBM-2R: 5′-AATGTGGTGTTGAGGTTGCGGTCTGTT-3′) based on the sequences of a sibling species, Red-billed Blue Magpie (U. erythrorhyncha; Genbank accession no. JQ423932). Two parts of long mtDNA sequence were sequenced separately by primer walking on an ABI 3730 DNA Analyzer (Applied Biosystems, Foster, CA). It took 26 primer pairs to complete the primer walking procedure, and the sequenced fragments were assembled into the whole mitochondrial genome sequence with the aid of SeqMan version 7.1 (DNAStar, Madison, WI) and then adjusted manually by eye. The whole mitochondrial genome sequence was deposited in GenBank with the accession number MG932654.
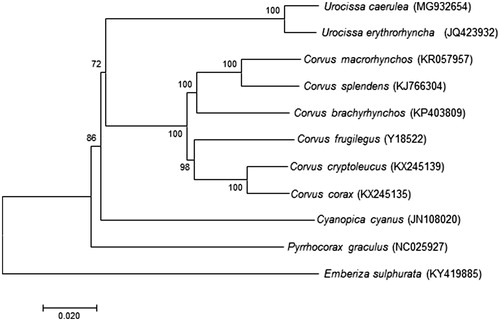
The organization of mitochondrial genome of D1818 is determined to be 16,928 bp in size, including 13 typical vertebrate protein-coding genes, 22 transfer RNA genes, 2 ribosomal RNA genes, and a control region (CR). All genes were encoded on the H-strand with the exception of one protein-coding gene (ND6) and eight tRNA genes [tRNAGln, tRNAAla, tRNAAsn, tRNACys, tRNATyr, tRNASer(UCN), tRNAPro, and tRNAGlu]. The base composition was counted using MEGA7 (Kumar et al. Citation2016). The overall base composition in descending order is: A (30.99%), T (24.697%), C (30.07%), G (14.25%) with 44.32% GC content. The positions of RNA genes were predicted by the MITOS (Bernt et al. Citation2013) and the locations of protein-coding genes were identified by comparing with the homologous gene of Red-billed Blue Magpie. The 22 tRNA genes range from 69 to 75 bp in length and all can fold into a typical cloverleaf secondary structure that was estimated by the online software tRNAscan-SE v2 (Lowe and Chan Citation2016). The two ribosomal RNA genes, 12S rRNA (947 bp) and 16S rRNA (1602 bp), were located between tRNAPhe and tRNALeu(UUR) genes and were separated by the tRNAVal gene as seen in other vertebrates. Except for COX1 and ND4L, the remaining 11 protein-coding genes start with an ATG codon. Eleven protein-coding genes end with complete stop codons TAA (ND2, COX2, ATP8, ATP6, ND3, ND4L, CYTB, and ND6), AGA (ND1 and ND5), and AGG (COX1). The remaining protein-coding genes end with the incomplete stop codons represented by ‘T’ (COX3 and ND4). CR is 1347 bp long. No tandem repeat segment was found, checked by online software ‘TANDEM REPEATS FINDER’ (Benson Citation1999). Similar to other Aves’ mitochondrial genome organization, the gene arrangement between ND5 and CR is CYTB- tRNAThr- tRNAPro- ND6- tRNAGlu (e.g. Härlid et al. Citation1998; Lee et al. Citation2017; Ludwig et al. Citation2017), which is different from that of mammals and fish (ND6- tRNAGlu -CYTB- tRNAThr- tRNAPro; e.g. Xu et al. Citation1996; Chang et al. Citation2013, Citation2016; Jang-Liaw et al. Citation2013; Chiang et al. Citation2017; Hou et al. Citation2018), as well as the absence of the origin of L-strand replication (OL), which is common in other vertebrates’ mitochondrial genome. As shown in the Neighbor-Joining (NJ) analysis, which included mitochondrial genome of U. caerulea and the other 10 species from the family Corvidae using Emberiza sulphurata (Passeriformes) of family Fringillidae as an outgroup (), U. caerulea clustered with another Urocissa species, U. erythrorhyncha, and is highly supported by a bootstrap value of 100. Thus, the newly determined mitochondrial genome will enrich the basic molecular data of Urocissa.
Figure 1. Neighbor-Joining (NJ) phylogenetic tree of U. caerulea and the other 10 species using Emberiza sulphurata as an outgroup were conducted in MEGA7. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1000 replicates) are shown next to the branches. The evolutionary distances were computed using the Kimura 2-parameter method and are in the units of the number of base substitutions per site. All species’ accession numbers are listed behind taxa.
Acknowledgements
We are grateful to Tedd Chen for editing assistance. This work was supported by 2017 Summer Intern Student Project of Taipei Zoo.
Disclosure statement
No potential conflict of interest was reported by the authors.
Related Research Data
References
- BirdLife International. 2016. Urocissa xanthomelana. The IUCN Red List of Threatened Species 2016: e.T103719397A104096516. [accessed 2018 Feb 13]. http://dx.doi.org/10.2305/ IUCN.UK.2016-3.RLTS.T103719397A-104096516.en
- Benson G. 1999. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27:573–580.
- Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G, Pütz J, Middendorf M, Stadler PF. 2013. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 69:313–319.
- Chang CH, Jang-Liaw NH, Lin YS, Carlisle A, Hsu HH, Liao YC, Shao KT. 2016. The complete mitochondrial genome of the salmon shark, Lamna ditropis (Chondrichthyes, Lamnidae). Mitochondrial DNA A DNA Mapp Seq Anal. 27:316–317.
- Chang CH, Tsai CL, Jang-Liaw NH. 2013. Complete mitochondrial genome of the Chinese rasbora Rasbora steineri (Teleostei, Cyprinidae). Mitochondrial DNA. 24:183–185.
- Chiang WC, Chang CH, Hsu HH, Jang-Liaw NH. 2017. Complete mitochondrial genome sequence for the green humphead parrotfish Bolbometopon muricatum. Conservation Genet Resour. 9:393–396.
- Härlid A, Janke A, Arnason U. 1998. The complete mitochondrial genome of Rhea americana and early avian divergences. J Mol Evol. 46:669–679.
- Hou HY, Chang RX, Cheng YN, Jang-Liaw NH. 2018. Complete mitochondrial genome sequence for the somali wild ass Equus africanus somaliensis. Conserv Genet Resour. https://doi.org/https://doi.org/10.1007/s12686-018-1035-y
- Integrated Taxonomic Information System. 2018. ITIS Report: Urocissa. [accessed 2018 Feb 14].
- Jang-Liaw NH, Chang CH, Tsai CL. 2013. Complete mitogenomes of two Puntius in Taiwan: P. semifasciolatus and P. snyderi (Cypriniformes: Cyprinidae). Mitochondrial DNA. 24:228–230.
- Kumar S, Stecher G, Tamura K. 2016. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 33:1870–1874.
- Lee MY, Jeon HS, An J. 2017. Complete mitochondrial genome sequence of Emberiza sulphurata (Emberizidae: Emberiza). Mitochondrial DNA B Resour. 2:111–112.
- Lowe TM, Chan PP. 2016. tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 44:W54–W57.
- Ludwig S, Martins APV, Queiroz ALL, Carmo AOd, Oliveira-Mendes BBR, Kalapothakis E. 2017. Complete mitochondrial genome of Sporophila maximiliani (Ave, Passeriformes). Mitochondrial DNA B Resour. 2:417–418.
- Xu X, Gullberg A, Arnason U. 1996. The complete mitochondrial DNA (mtDNA) of the donkey and mtDNA comparisons among four closely related mammalian species-pairs. J Mol Evol. 43:438–446.