
Abstract
The complete mitogenome of Caryanda elegans is 15,459 bp in length and consists of 13 protein-coding genes, 22 tRNA genes, 2 rRNA genes and 1 A + T-rich region. The gene order of the mitogenome is identical with most orthopteran insects. Total 105 bp intergenic spacers (IGSs) in 19 regions and total 49 bp overlaps in 9 regions were found in this species. The As, Ts, Cs, Gs and AT content of the mitogenome is 42.8%, 32.1%, 14.5%, 10.6% and 74.9%, respectively. All protein-coding genes start with typical ATN codon, and end with TAA codon except for ND5 and ND1, which terminated with T and TAG codons instead, respectively. Phylogenetic analysis indicated that genetic distances of C. elegans and Pseudoxya diminuta was closer than other species.
Keywords:
The genus Caryanda (Orthoptera: Caelifera: Acrididae) consists of 64 species, mainly distributed in the southeast of Asia and central of Africa. Caryanda elegans, small size and yellowish green body, which usually lives in the dense grass of hillside, especially lives in the lush grass under trees and large rocks. This insect is mainly distributed in Yunnan, Sichuan and Guangxi provinces in China (Jiang and Zheng Citation1998) and Tinh Lào Cai in Vietnam (Kim and Pham Citation2014). C. elegans is the most widely distributed species while the other species within this genus are more strictly localized. At present, data on the biology were scarce, especially, the limited molecular data dampen the evolution and diversity studies in genus Caryanda.
The specimen of C. elegans was collected from Meng Yang County, Yunnan Province, China, and was deposited in the animal herbarium of Shaanxi Normal University. Genomic DNA was prepared in paired-end libraries, tagged and subjected to the high-throughput Illumina Xten platform with 150 bp paired-end strategy and yield 7,962,577 Paired-End Raw Reads. Clean reads were trimmed by removing regions with a Phred score of <10. High-quality reads were assembled using MITObim version 1.9 (Hahn et al. Citation2013) and the complete mitogenome of Oxya chinensis (GenBank: NC_010219) as a reference. A total of 15,764 individual mitochondrial reads gave an average coverage of 160.4X. Comparing with the O. chinensis, annotations were generated in Geneious version 10.1.2.
The complete mitogenome of C. elegans is 15,459 bp in length (GenBank: MF095791), including 13 protein-coding genes, 22 tRNA genes, 2 rRNA genes and 1 A + T-rich region. The As, Ts, Cs and Gs content of the mitogenome was 42.8%, 32.1%, 14.5% and 10.6%, respectively, and the AT content is 74.9%. The tRNA genes were predicted by online software MITOchondrial genome annotation Server (MITOS) (Bernt et al. Citation2013), and the length ranked from 64 bp (tRNA-Cys and tRNA-Pro) to 71 bp (tRNA-Val and tRNA-Asp). There are a total of 49 bp overlaps in the mitogenome of C. elegans at nine locations with the length ranging from 1 bp to 10 bp, and the longest overlaps occur between ND2 and tRNA-Trp. Total 105 bp were found in 19 intergenic spacers in this mitogenome with the length ranging from 1 to 20 bp, and the longest one was 20 bp between tRNA-Ser(UCN) and ND1. All tRNAs could be folded into typical cloverleaf secondary structures, except tRNA-Ser(AGN), whose dihydrouridine arm forms a simple loop as in most other insects (Ohtsuki et al. Citation2002). The length of 12S rRNA was 843 bp and 16S rRNA was 1315 bp, located between tRNA-Leu(CUN) and A + T-rich region, separated by tRNA-Val. The typical ATN start codon was presented in 13 PCGs, and most PCGs of C. elegans end with TAA, while ND1 ends with TAG, ND5 ends with the incomplete stop codon T. The A + T-rich region, which is located between 12S rRNA and tRNA-Ile, is 577 bp in length and has 85.1% AT content.
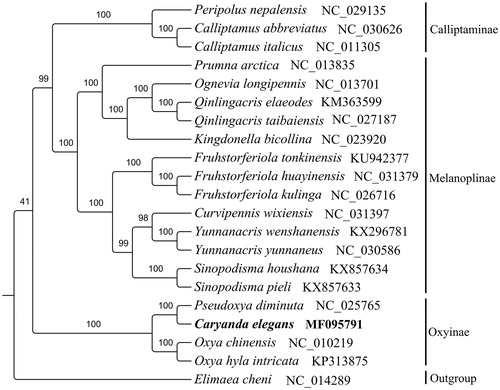
To further validate the mitogenome of C. elegans, a phylogenetic analysis was built based on 13 PCGs and 2 rRNA genes with 20 ingroups and 1 outgroup (). The result shows that C. elegans can be unambiguously grouped in Oxyinae with high bootstrap value supporting. C. elegans and Pseudoxya diminuta are sister group. Totally, the mitogenome of C. elegans provides essential and important DNA molecular data for further phylogenetic and evolutionary analysis for Oxyinae.
Figure 1. Maximum likelihood tree obtained using RaxML (Stamatakis Citation2014) with 1000 non-parametric bootstrap replicates. DNA sequences were aligned with MEGA4.0 (Tamura et al. Citation2007) individually and alignments of individual genes were concatenated using SequenceMatrix v1.7.8 (Vaidya et al. Citation2011). PartitionFinderV1.1.1 (Lanfear et al. Citation2012) was used to determine the optional model of evolution and partitioning scheme simultaneously. All mitogenomes were downloaded from GenBank except C. elegans, and the accession number was given with species names.
Disclosure statement
The authors declare no competing interests.
Additional information
Funding
References
- Bernt M, Donath A, Jühling F. 2013. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 69:313–319.
- Hahn C, Bachmann L, Chevreux B. 2013. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads—a baiting and iterative mapping approach. Nucl Acids Res. 41:e129–e129.
- Jiang GF, Zheng ZM. 1998. Grasshoppers and locusts from Guangxi. Guilin: Guangxi Normal University Press; p. 1–400.
- Kim TW, Pham HT. 2014. Checklist of Vietnamese orthoptera (Saltatoria). Zootaxa. 3811:53–82.
- Lanfear R, Calcott B, Ho SYW, et al. 2012. PartitionFinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol Biol Evol. 29:1695–1701.
- Ohtsuki T, Kawai G, Watanabe K. 2002. The minimal tRNA: unique structure of Ascaris suum mitochondrial tRNA(Ser)(UCU) having a short T arm and lacking the entire D arm. FEBS Lett. 514:37–43.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30:1312–1313.
- Tamura K, Dudley J, Nei M, et al. 2007. MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol. 24:1596–1599.
- Vaidya G, Lohman DJ, Meier R. 2011. SequenceMatrix: concatenation software for the fast assembly of multi‐gene datasets with character set and codon information. Cladistics. 27:171–180.