
Abstract
Caranx equula is an important marine fish which due to its commercial values. However, some wild populations of C. equula are in danger because of the overfishing and environmental pollution. In this study, the complete mitochondrial genome of C. equula was firstly determined. The complete mitochondrial genome is 16,607 base pairs in length, and contained 13 protein-coding genes and 2 ribosomal RNA genes, 22 tRNA genes and 2 main non-coding regions. Overall A + T content was 52.9%. In addition, a phylogenetic tree was constructed using the complete mitochondrial genome and showed that C. equula clustered in a clade and formed a sister relationship with Caranx ignobilis belonged to the family of Carangidae.
Caranx equula belongs to the family Carangidae, which is widely distributed in tropical waters of Indo-Pacific. It is a reef-associated fish and famous for its edibleness and economic value. Although it has these significant values, many of its wild populations are in danger due to the ecological damages, overfishing, and marine pollution. Now the conservation of C. equula need to be pay an important attention. In this study, the complete nucleotide sequences for the mitochondrial genome of the C. equula was present. And to examine the evolutionary position of C. equula, a phylogenetic tree using Bayesian inference analysis was constructed. The present result will facilitate further investigations on the taxonomic resolution and genetic conservation of Carangidae.
The specimen of C. equula was collected from the waters around the East China Sea (29.9'N, 122.2'E) and was kept at the Museum of Marine Biology at Zhejiang Ocean University. Eight primer pairs were designed to amplify the complete mitochondrial genome of C. equula (Supplemental Table S1). The sequence fragments were assembled. The tRNA genes were identified by their proposed clover-leaf secondary structure and anti-codons. All the sequences collected from GenBank (Supplemental Table S2) were used to construct phylogenetic trees by Bayesian inference analysis.
The mitochondrial DNA of C. equula is a closed double-stranded circular molecule of 16,607 bp (GenBank accession number: KX373635) and contained 13 protein genes, 2 rRNA genes, 22 tRNA genes, 1 putative control region (CR), and 1 light strand replication origin (OL; Supplemental Table S3). The base composition of the complete mitochondrial genome was A 27.9%, C 30.4%, G 16.7%, and T 25%, with a slight A + T bias of 52.9% (Supplemental Table S4), which was similar to other fishes mitochondrial base content (Liu et al. Citation2013; Cheng et al. Citation2012). The 13 protein-coding genes had a strong bias against G (8.4%) at the third codon position, which was typical in vertebrate mitochondrial genomes (Ishiguro et al. Citation2001). Most of these genes are encoded on the H-strand, except for the ND6 gene and eight tRNA genes (ND6, tRNAGln, tRNAAla, tRNAAsn, tRNACys, tRNATyr, tRNASer, tRNAGlu, and tRNAPro), which are encoded on the L-strand.
The 12S and 16S rRNA genes of C. equula which are similar to other vertebrates, located between tRNAPhe and tRNALeu (UUR) genes, and are separated by the tRNAPhy. All the protein-coding genes started with ATG, except for COI used GTG, ATP6 used ATA as the start codon. The stop codon of seven protein-coding genes (ND1, COI, ATP8, ATP6, ND4L, ND5, and ND6) is TAA. The remaining four genes seemed to end in two incomplete stop codons, TA- (COIII) and T- (ND2, COII,ND3, ND4, and Cytb), which were completed via post-transcriptional polyadenylation (Ojala et al. 1981).
The 22 tRNA genes which have two forms, including tRNASer (UCN and AGY) and tRNALeu(UUR and CUN), scatter throughout the genome and range from 57 to 74 bp in size and the gene arrangement is typically as in most vertebrates. Three tRNA clusters (IQM, HSL, and WANCY) were well conserved in C. equula as in other vertebrate mitochondrial genomes. Fourteen tRNA genes were encoded by H-strand, while the remaining eight tRNA genes (tRNAGln, tRNAAla, tRNAAsn, tRNACys, tRNATyr, tRNASer, tRNAGlu, and tRNAPro) were encoded by L-strand. The total length of overlaps and intergenic spacers were 18 and 82 bp, ranging from 1 to 7 bp and from 1 to 36 bp per location, respectively. As in most vertebrates, two non-coding regions can be found in C. equula mitogenome, an OL within the WANCY region including five tRNA genes (tRNATrp, tRNAAla, tRNAAsn, tRNACys, and tRNATyr), which can fold into a stem loop secondary structure with the conserved motif 5′-GCCGG-3′. The control region in C. equula mitogenome is determined between tRNAPro and tRNAPhe and this characterization is consistent with those of other teleost (Jin et al. Citation2013; Wei et al. Citation2013; Xu et al. Citation2011).
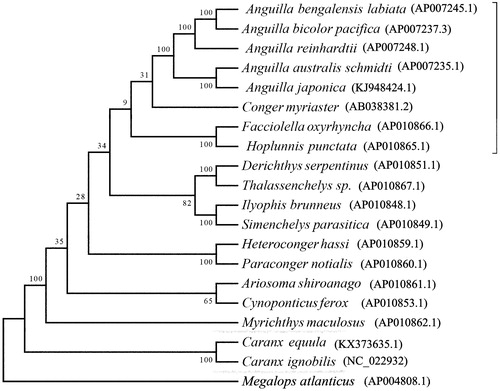
To investigate the phylogenetic position of C. equula among closely related fishes, a phylogenetic tree was constructed with other teleost mitochondrial genome sequences using Bayesian inference analysis. The mitochondrial genome sequence of Megalops atlanticus was used as outgroup. Phylogenetic analysis result demonstrated that C. equula clustered in a clade and formed a sister relationship with other species belong to the family of Carangidae (). This complete mitochondrial genome can be used for population genomic studies and the data will provide fundamental information for the genetic conservation and the taxonomic resolution of Carangidae.
Figure 1. Phylogenetic tree based on the complete mitochondrial genome sequences was constructed by using Bayesian method. The numbers in topologies represent Bayesian posterior probability values.
Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the manuscript.
References
- Cheng YZ, Wang RX, Sun YN, Xu TJ. 2012. The complete mitochondrial genome of the small yellow croaker and partitioned Bayesian analysis of Sciaenidae fish phylogeny. Genet Mol Biol. 35:191–199.
- Ishiguro N, Miya M, Nishida M. 2001. Complete mitochondrialDNA sequence of ayu Pleoglossus altivelis. Fisheries Sci. 67:474–481.
- Jin XX, Gao YH, Xu TJ, Shi G, Zhao SL, Sun YN. 2013. Complete mitochondrial genome of the Asian freshwater goby Synechogobius ommaturus (Perciformes, Gobioidei). Mitochondrial DNA. 24:83–85.
- Liu TX, Jin XX, Wang RX, Xu TJ. 2013. Complete sequence of the mitochondrial genome of Odontamblyopus rubicundus (Perciformes: Gobiidae): Genome characterization and phylogenetic analyses. J Genet. 92:423–432.
- Ojala D, Montoya J, Attardi G. 1981. tRNA punctuation model of RNA processing in human mitochondria. Nature. 290:470–474.
- Wei T, Jin XX, Xu TJ. 2013. The first complete mitochondrial genome from Bostrychus Genus (Bostrychus sinensis) and partitioned Bayesian analysis of Eleotridae fish phylogeny. J Genet. 92:247–257.
- Xu TJ, Chen YZ, Liu XZ, Shi G, Wang RX. 2011. The complete mitochondrial genome of the marbled rockfish Sebastiscus marmoratus (Scorpaeniformes, Scorpaenidae): genome characterization and phylogenetic considerations. Mol Biol. 45:392–403.